Article Text

Abstract
Background Technology, equipment and medical devices are vital for effective healthcare throughout the world but are associated with risks. These risks include device failure, inappropriate use, insufficient user-training and inadequate inspection and maintenance. Further risks within the developing world include challenging conditions of temperature and humidity, poor infrastructure, poorly trained service providers, limited resources and supervision, and inappropriately complex equipment being supplied without backup training for its use or maintenance.
Methods This document is the product of an expert working group established by WHO Patient Safety to define the measures being taken to reduce these risks. It considers how the provision of safer technology services worldwide is being enhanced in three ways: through non-punitive and open reporting systems of technology-related adverse events and near-misses, with classification and investigation; through healthcare quality assessment, accreditation and certification; and by the investigation of how appropriate design and an understanding of the conditions of use and associated human factors can improve patient safety.
Results and discussion Many aspects of these steps remain aspirational for developing countries, where highly disparate needs and a vast range of technology-related problems exist. Here, much greater emphasis must be placed on failsafe, durable and user-friendly design—examples of which are described.
- Safety
- safety management
- technology
- risk management
- quality of healthcare
- adverse event
- human factors
- incident reporting
- safety culture
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Safety
- safety management
- technology
- risk management
- quality of healthcare
- adverse event
- human factors
- incident reporting
- safety culture
Introduction
The ubiquity and usage of equipment and technology within healthcare are growing rapidly, with over US$130 billion spent in the USA alone in 2006 on medical devices.1 Although essential for advances in modern medicine, many established and associated risks of technology continue.2 It is therefore paramount to reduce the potential risk using a combination of methods that link human factors, equipment and the healthcare environment, as shown in figure 1.

Reducing the risk associated with technology within healthcare. Mechanisms for addressing and reducing risk associated with technology in healthcare.
A WHO Patient Safety working group was established to consider how existing technology can be made safer. The group includes representatives from high-, middle- and low-income countries with expertise in clinical medicine, academia, policy, health services management and industry. It is guided by a panel of international experts and draws on the scientific literature, where available, that is associated with the safety of current technology in the healthcare environment. Educational bodies and health service providers were approached to provide information on the specific technology problems that developing countries face. This report on this work is global in its scope, considering both the developed and developing world.
We have used the definition of ‘Health Technology’ adopted by the Health Technology Assessment programme in the UK—‘a range of methods used to promote health, prevent and treat disease and improve rehabilitation and long-term care, including drugs, devices, procedures, settings of care and screening’—but have avoided any analysis of pharmacovigilance efficacy. One paper within the supplement recommends an agenda for future research within the field, whereas another outlines how new technology can be introduced safely.
We have identified four broad themes:
The importance of reporting and learning systems to identify areas where technology is unsafe—importantly, these demonstrate that even in equipment-rich environments, such as critical care and anaesthetics, fewer than one in 10 incidents of healthcare-associated harm or death are attributable to actual device failure or faults.3 4
Establishing systems of healthcare accreditation to ensure continuous evaluation and quality improvement.
Because the majority of adverse incidents are associated with improper use and problems at the interface between equipment, users and patients, greater consideration needs to be given to human factors5 and intelligent redesign.
The specific challenges and issues in developing countries.
Adverse incident reporting
Reporting systems provide a mechanism for enhancing patient safety through learning from failures reported by healthcare workers. They reflect a measure of progress towards achieving a safety culture. The primary purpose of reporting systems for adverse incidents and near-misses within healthcare is to learn from experience.6 However, reporting systems do not improve safety directly. It is the analysis of reports and subsequent dissemination and implementation of recommendations (eg, announcing recalls and safety alerts)4 that leads to changes. Serious incident reports should trigger an extensive investigation to identify underlying systems failures and lead to efforts to redesign the systems to prevent recurrences. Although most incident reporting systems suffer from under-reporting for a variety of reasons,7 8 and are restricted by a lack of denominator data, there are several ways in which reporting can lead to learning and improved safety.
Early warning systems for device failure: These can generate alerts regarding new and unsuspected hazards, and ‘accidents waiting to happen,’ as a means of achieving prevention without the need to learn from an injury.9 This could result from a few similar incoming reports picked up by human review of previously unrecognised complications associated with the use of a new device. For example, even if only a few people report that free-flow protection on a particular pump model can fail, that might be sufficient for the receivers of the reports to recognise the problem, alert the providers and communicate directly with the pump manufacturer.
Early warning systems for poor device design: Reporting could identify an important gap in current safety systems, such as devices with designs or interfaces that allow or induce misuse in ways that can produce serious adverse events—even though they still meet the manufacturer's specifications and pass regulatory standards. Conversely, a well-known example of surveillance failure is the software bugs of the Therac-25 linear accelerator for radiation therapy during the mid-1980s.10 Inadequate reporting mechanisms and communication between hospitals, the Food and Drug Administration (FDA) and manufacturer were partly responsible for ongoing fatal radiation overdoses in six patients over a period of 18 months.
Detecting problems that occur after several years: Some problems are not highlighted during short premarket studies and take years to become apparent.
Detecting problems that rarely occur: By summing a large number of reports, it is possible to detect rare problems or complications that would not be detected by premarketing studies of limited size. This is the major thinking behind pharmacovigilance systems.11
Opportunity for analysis: Analysis of many reports by the receiving agency or others can reveal unrecognised trends and hazards requiring attention. Analysis of multiple reports can lead to insights into underlying systems failures or specific patient factors associated with technologically related adverse events.12 13 This generates both priority areas for remedial efforts and educational recommendations for ‘best practices.’
Surveillance/reporting methods
There is no ‘gold standard’ for the surveillance or identification of Medical Device Related Events (MDREs) or their subsequent reporting.5 Samore et al compared six methods for exclusively highlighting MDREs, in a US tertiary teaching hospital in 2000. Importantly, they found minimal overlap in the events identified by the different methods.14 During 20 441 inpatient stays, an online incident reporting system voluntarily completed by healthcare professionals highlighted only 80 MDREs, whereas 1359 reports were logged to the hospital's clinical engineering department. During the 9-month study, 1122 International Classification of Diseases, Ninth Revision (ICD-9) MDRE-related codes were ascribed by the hospital's administration at patient discharge, and a postdischarge patient survey found that 7% of patients considered that there had been problems with medical devices during their stay. A voluntary telemetry checklist yielded no MDREs. This study found that automated surveillance of the electronic medical record (previously shown to detect adverse drug events)15 using seven selected ‘flags’ had a 7.8% positive predictive value (PPV), with only 552 out of 7059 ‘flagged’ events being actual MDREs.
By using an example of an incident-management system based on a universal classification system, 43 desirable attributes of an integrated framework for safety, quality and risk management have been described previously.9 Once this classification can be agreed internationally (see below), work could proceed under the auspices of WHO Patient Safety on developing standards, field formats for data collection, aggregation, storage and analysis, and, ultimately, make it easy to allow data sharing and the creation of a universal database, as foreshadowed in 2002.13
International classification for patient safety
To date, incident reporting has been compromised by a lack of agreed definitions and preferred terms for the key concepts necessary to describe the attributes, characteristics, limitations and pitfalls of underlying healthcare technologies.16 17 To promote a common understanding and ease the comparison of international datasets from different reporting systems, WHO Patient Safety commissioned work to develop a framework for an International Classification for Patient Safety (ICPS) (figure 2).18 19

Conceptual framework for the International Classification for Patient Safety (ICPS). Maximum information collected from all adverse events and near misses is grouped into incident types (‘medical device/equipment/property’ and ‘infrastructure/building/fixtures’ having direct relevance to technology). During the incident, mitigating factors prevent or moderate harm to the patient. Organisational outcomes refer to the effects on the organisation, such as appropriation of resources to the affected patient. Adapted from Sherman et al.19
Sources of information for the ICPS include incident reports, medicolegal files, coroners' recommendations, complaints and audits. Clinical engineering departments also use failure-related data extracted from computerised medical-equipment management systems for risk-management purposes. Reporting via a call centre equipped with the appropriate software for eliciting the information needed to populate a classification such as the ICPS would be cost-effective and require little infrastructure. Such a system is being used successfully in South Africa (see box 1). The database is populated in English using operators who speak English as well as the language of the reporter.20
Box 1 How the safety of technology in developing world healthcare can benefit from incident reporting systems
Incident reporting in South Africa
In South Africa, COHSASA is piloting the Advanced Incident Management System (AIMS) previously developed in Australia. Healthcare staff report problems via telephone to a call centre where data are recorded onto the AIMS database. These problems are then analysed and a report is quickly sent back to the institution. AIMS also assesses the institution's response to the report. In South Africa, insufficient consumables have been immediately evident as frequent causes of adverse events that are quickly rectifiable. For example, the absence of Yankauer suction tubing led to the death of a patient because it was not possible to perform tracheal suction. Similarly, an unexpected neonatal death occurred because of a failure to recognise the deterioration of simple physiological parameters in a newborn because there were insufficient pulse oximeters. (Case reports provided by COHSASA.)
Collecting information from all available sources into a classification system such as the ICPS can help to identify problems early. Clinicians and regulatory authorities in that jurisdiction, as well as in other countries, can then be alerted.
Importance of transparent non-punitive reporting
To encourage reporting and an appropriate response within the prevalent ‘blame culture,’ successful patient safety reporting systems must employ a non-punitive ‘just culture’ approach21 22 except in the case of significant negligence. Neither reporters nor others involved in the incidents should be punished as a result of reporting, with the knowledge that adverse events and errors are symptoms of defective systems, not defects themselves. Figure 3 shows the sharp rise in telephone reporting to the Advanced Incident Management System (AIMS) in South Africa's North West Hospitals with a just culture approach in which staff were given the written assurance that they would not be punished regarding adverse incident reports. This excluded staff that displayed reckless behaviour by ignoring well-known safety protocols but included human error and at-risk behaviour (staff who did not know that they were doing wrong). Many systems offer the option of reporter anonymity, which increases the rate of reporting.20 23

How a ‘just culture’ improves reporting: the rise in telephone reporting of adverse incidents to the Advanced Incident Management System in South Africa's North West Hospitals after a guarantee to staff of a non-punitive just culture during the first 10 months of 2008. Adapted from data provided by the Council for Health Service Accreditation of Southern Africa.
Types of reporting systems
The most modest reporting systems are local audit. For example, a recent Tanzanian audit suggested that a quarter of perinatal deaths were associated with inadequate maternal and fetal heart monitoring.24 If the audit cycle is completed, awareness of these deficiencies leads to improved safety.25
Generally, reporting systems can be either mandatory or voluntary and either held in complete confidence or reported to the public or to regulatory agencies. Reporting systems are generally internal or external and are open-ended, capturing all adverse events across care delivery, or focus on particular types of events such as predefined serious injuries, epidemiological outcomes such as the emergence of antimicrobial resistance or blood transfusion events. An example of the latter is the UK's Serious Hazards of Transfusion (SHOT) organisation, which was established in 1996 to encourage all hospitals in the UK to participate in haemovigilance to enable the identification and dissemination of solutions to make transfusion safer. Since its inception, SHOT has borne the hallmarks of an effective vigilance system with rising reporting accompanied by a steady decline in transfusion-associated mortality in the UK.26
Formats and processes within different reporting systems vary from prescribed forms with defined data elements to free-text reporting. It is imperative that sufficient information is provided for subsequent analysis—for example, the make and model of ventilator.4 The system might allow for reports to be submitted in various formats including mail or telephone,5 although electronic submission is arguably easier27 and becoming more commonplace.
Some systems primarily have learning objectives, for example for device reporting to the FDA,28 29 whereas others are designed to provide accountability. Rather than ensure a minimum standard of care, learning systems are designed to foster continuous improvements in care delivery by identifying themes in adverse events and near-misses, reducing variation in their incidence, facilitating the sharing of best practices and stimulating system-wide improvements. Incident reporting within learning systems is usually voluntary, and, via careful expert analysis of the underlying root causes, recommendations are made to redesign and improve the performance of systems in order to reduce errors and injuries. For example, the National Reporting and Learning System (NRLS) in England and Wales receives reports of patient safety incidents from local healthcare organisations. Its annual summary in November 2008 found that 27% of the 656 781 inhospital reports were about problems arising from medical devices or equipment.30 About 1% of these caused death or severe harm to the patient.31
Conversely, reporting in accountability systems is usually mandatory and restricted to a list of defined serious events (also called ‘sentinel’ events) such as unexpected death, transfusion reaction and surgery on the wrong body part. Accountability systems typically prompt improvements by requiring an investigation and systems analysis (‘root cause analysis’) of the event. However, few regulatory agencies have the resources to perform external investigations of more than a small fraction of reported events, which limits their capacity to learn. Table 1 gives further examples of both systems.
Further examples and descriptions of both learning and accountability types of reporting systems
Most accountability systems hold healthcare organisations accountable by requiring that serious mishaps be reported. Furthermore, they provide disincentives to unsafe care through citations, penalties or sanctions. The effectiveness of these systems depends on the ability of the agency to induce healthcare organisations to report serious events and to conduct thorough investigations.
For any system, the analysis of reports with assessment of risk needs to be prompt, with notification of serious hazards being made without delay. With a large number of reports, estimations of the probability of recurrence of a specific type of adverse event or error can be calculated. Analysis of reported outcomes can also produce an estimate of the average severity of harm caused by the incident or type of incident.34 Risk analysis should be carried out by the most appropriate committees found within the healthcare facility. Depending on the institution, this might include an advisory committee on healthcare technology, the resuscitation committee, a health and safety committee or a theatre-users committee. Findings from reporting systems inform new safety initiatives that are generated and implemented by the appropriate authority. For example, the suggestion that adequate monitoring with capnography and oximetry would have resulted in the detection of 88% of the first 2000 anaesthesia-related adverse events reported to AIMS in Australia35 had a major impact on the International Standard for Anaesthesia Safety that was endorsed in 1994.36
Unfortunately, the national and international reporting and surveillance systems that exist in developed countries are scarce or new in developing countries (box 1), and little is known about the frequency or impact of events involving medical devices.
Healthcare quality assessment, accreditation and certification
Evaluating, certifying and monitoring the quality of the provision of healthcare services using agreed standards is an excellent method of improving the safety of healthcare technology, particularly when it prompts change, subsequent reappraisal and a culture of continuous improvement, problem solving and critical self-examination. Quality assurance and improvement are achieved by ensuring standards of governance, using performance measures or indicators to measure an organisation's performance and encouraging the use of guidelines. Accreditation sceptics cite an increased workload, particularly for hospital middle management, a lack of consistency and significant cost. With reference to technology, however, accreditation can encourage training and continued professional development, improve audit and catalyse change to equipment and estates.37 Examples include the Joint Commission and the Community Health Accreditation Program (CHAP) from the USA, the Trent Accreditation Scheme (TAS) in the UK and the Australian Council on Healthcare Standards.
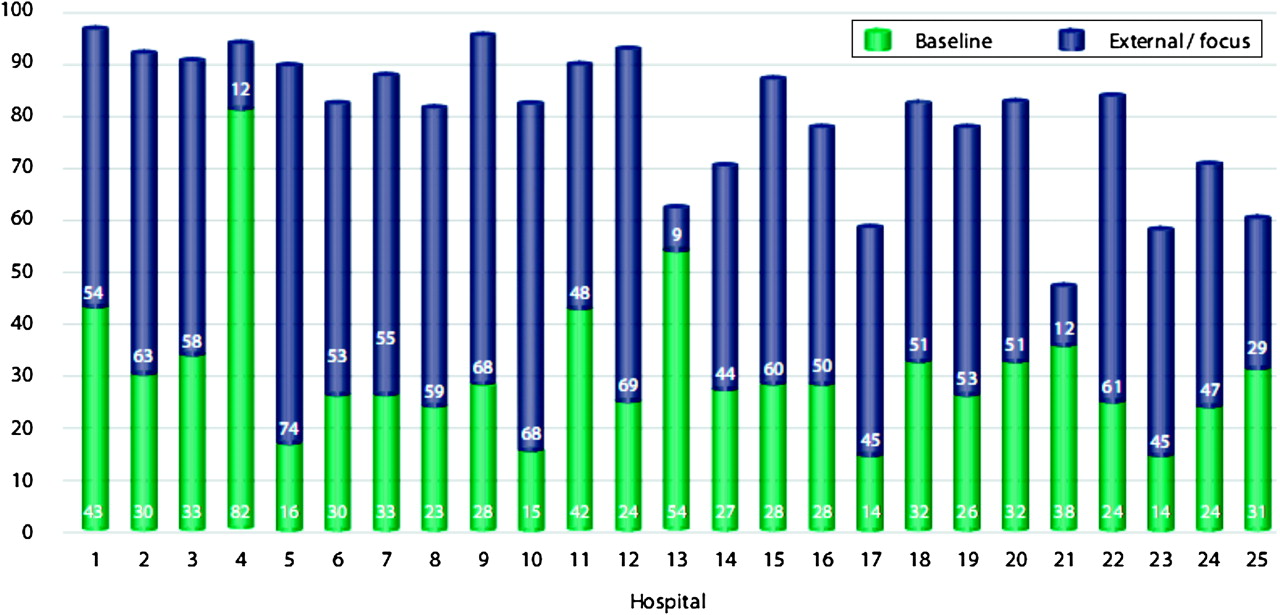
Quality assurance is possible in the developing world:38 39 in South Africa, COHSASA uses standards that define the key functions, activities, processes and structures required for the health facility departments to be in a position to provide quality Healthcare Technology Management services (HTM) that meet the principles set out by the International Society for Quality in Health Care (ISQua).40 Accreditation is provided if minimum standards are demonstrated across seven areas: medical equipment support, healthcare technology planning, policies and procedures, medical equipment management, staff training, quality improvement and equipment safety. The ‘equipment safety’ area assesses the institutions' risk management and performance testing services, as well as the safety of the working conditions for the staff and their involvement in electrical safety training. In a typical programme, a baseline survey of an entire hospital is undertaken. Areas of non-compliance are identified, which for COHSASA are more commonly HTM planning, equipment safety and quality improvement. A multidisciplinary, continuous quality improvement approach follows, and external surveys are carried out by peers at various stages during the process. Figure 4 shows how mean levels of compliance across COHSASAs seven areas of HTM can be improved.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Healthcare Technology Management (HTM) scores for 25 facilities at baseline and after quality improvement. The mean performance indicator scores were measured over 5 years for the seven HTM areas at baseline (green bar) and after quality improvement at least 18 months later (blue bar) in 25 different institutions in South Africa. Scores greater than 80 are acceptable, and scores under 40 are considered highly unsatisfactory. Adapted from Council for Health Service Accreditation of Southern Africa data.
A culture of quality and safety has also been encouraged in Ghana with the establishment of a Non-Governmental Organisation (NGO) called the Ghana Quality Organisation collaborating with the Ghana Health Service to launch a series of workshops, seminars and conferences.41
Certification of technological products within healthcare
Medical product regulation and certification relies heavily on the use of agreed standards from international NGOs such as the International Organisation for Standardisation42 (ISO with 163 member countries) and the International Electrotechnical Commission (IEC with 56 member countries). Standards include common safety symbols, common nomenclature and common paths for the validation of the safe usability of medical devices. The standards are nevertheless intended to allow individual manufacturers freedom to design their own solutions. The use of standards has given a strengthened focus on safety issues through safety-oriented standards such as quality-management systems and risk management applied to medical devices.43–45 However, this certification is more common in industrialised countries with existing regulatory frameworks. Furthermore, certification bodies are usually based in these richer countries, despite commendable efforts from standardisation bodies to increase stakeholder representation from all geographical areas and interest groups. Many advocate a widened stakeholder base in standardisation work. This would not only encompass the medical device industry, but also require more input from healthcare professionals, safety professionals and regulatory agencies—the latter providing invaluable information from adverse event reporting and postmarket surveillance systems.
Error, human factors and systems design
Human error
An error has been defined by WHO Patient Safety as ‘failure to carry out a planned action as intended or application of an incorrect plan.’18 Within the context of medicine, an adverse event is defined as ‘an unintended harm caused by medical management, rather than by a disease process, serious enough to lead to prolonged hospital admission, temporary or permanent disability to the patient.’46 James Reason has divided the investigation of human error into the person approach or the system approach.47 The person approach focuses on the errors of individuals, with blame for forgetfulness, inattention or moral weakness. This is rare, and only illustrated by high-profile cases such as those of the family doctor Harold Shipman48 and nurse Beverley Allit.49 The system approach focuses upon the work conditions, bringing with it the concept of ergonomics—the science of designing the job, equipment and workplace to fit the worker.
Reason states that latent conditions can provoke error in the workplace, through time pressures or inadequate staffing. Such conditions can lie dormant for long periods before they combine with active failures to produce an adverse event. Reason has famously proposed the ‘Swiss cheese model’ of error, whereby layers of swiss cheese act as defences to error.47 However, each layer has holes within it that are under a state of dynamic shift with regard to their presence, size and position. Each hole within a slice does not normally lead to a poor outcome, but when a number of holes in several layers line up, the potential for error production and propagation is great.
Vincent has expanded upon Reason's model to provide a classification of error-producing factors within a framework that can affect clinical practice.50 These range from task design and use of protocols, through to team communication and organisational structures. The report ‘To err is human’ was seminal in proposing that events causing or risking harm to patients were more likely to result from systemic failure,51 rather than the actions of individuals. The report suggested that efforts to improve patient safety should move away from a ‘blame culture’ and focus on removing ‘error-provoking’ aspects of care delivery systems. However, the traditional view of blaming and retraining an individual still prevails.52
Measurement of error
Measurement of error is difficult. Within the context of intervention, it is possible to produce a protocol that defines the steps in a prescribed order. Any deviation from this is defined as error. This approach, also known as Human Reliability Analysis, was explored by Joice et al during observation of laparoscopic surgical procedures.53 The aim was to define competent performance, although the study also demonstrated tasks that were more prone to error and instruments that were more likely to be associated with an error. The problem with such a tool is that it is quite focused upon the task in hand, and it is very difficult to consider the wider environment of the operating theatre with regards to patient factors, team factors and environmental factors.
Failure Modes and Effects Analysis (FMEA) is a procedure for analysis of potential failure modes within a system for classification by severity or determination of the effect of failures on the system.54 Failure modes are any errors or defects in a process, design or item, especially those that affect the customer (patient), and can be potential or actual. Effects analysis refers to studying the consequences of those failures. This tool has been hailed as a useful approach to identifying problems within wider healthcare processes, with a focus upon the interaction with technology. For example, FMEA has been used for analysis of processes of care related to medication delivery, infusion pumps, radiation therapy and suicide risk.54 Although a potentially useful tool, it is a laborious process requiring expert opinion upon the question in hand. Furthermore, its reliability has recently been questioned, with the conclusion that healthcare organisations should not depend solely upon FMEA findings to direct resources towards patient safety.55
Human factors
With particular focus on medical technologies, the aim is not only to produce high technology that serves a clinical purpose, such as a mechanical ventilator, but to ensure that the error-producing factors are considered with regards to placing such a device into clinical practice. There is of course a need to advance equipment design, but the importance of team structure and communication, organisational culture and crisis management cannot be understated. In high-reliability organisations such as the nuclear, oil and mining industries, these aspects are collectively known as human factors—a discipline that spans ergonomics, engineering and cognitive psychology. Human factor analysis focuses on performance design, incorporating human strengths and limitations, leading to iterative testing and evaluation. The importance of this concept to medicine is that testing occurs within an already functioning system (ie, in vivo) and could improve or endanger care.
The application of human factor approaches to medicine has been led by the anaesthetic community. In the 1970s, escalating litigation costs resulting from critical errors in anaesthesia led to analysis of near misses and fatal errors. This, in turn, led to the development of technologies to provide early warning of human or equipment error. Safety advances included non-interchangeable screw threads for different pipeline gases with the inlet on the anaesthetic machine, to prevent the delivery of hypoxic gases to the patient. Similarly, there has been work to develop separate ‘lock and key’ systems for intravenous and intrathecal delivery of medication—see the case study in box 2.
Box 2 Intelligent redesign resulting from a recurring severe adverse event
Vincristine: Wayne Jowett
Wayne Jowett was in remission from acute leukaemia, undergoing the final stages of his treatment. He was being treated with two chemotherapeutic drugs, vincristine given intravenously and cytosine given intrathecally. By mistake he was given vincristine intrathecally.56 Intrathecal vincristine causes paralysis and death. Wayne died a month after the injection. There have been over 50 cases of intrathecal vincristine reported worldwide.5 Despite awareness of the problem and repeated warning, this event still occurs. A variety of solutions have been proposed,57 including restrictions around seniority and training, separation of the intrathecally and intravenous drugs in time and space and technical solutions. Separate ‘lock and key’ systems for intravenous and intrathecal systems to prevent cross-use have long been viewed as the solution but have proved hard to achieve. More recently, the supply of vincristine in a ‘mini-bag’ of saline has been used. The volume of saline is such that no doctor or nurse would consider nor could inject the drug into the spinal space. However, owing to the volume of fluid, the mini-bags are not safe in the paediatric setting and so only represent a partial solution. These solutions represent key examples of redesigning technology to make care safer.
Systems design
To enhance patient safety, it is necessary to concentrate upon the systems approach to error and, in particular, upon latent failures. Solutions to such error and subsequent adverse events have been designed, investigated and implemented within medicine. The most recent and widely known is perhaps the WHO Surgical Safety Checklist project, which identifies three phases of an operation, each corresponding to a specific period in the normal flow of work: before the induction of anaesthesia (sign in); before the incision of the skin (time out); and before the patient leaves the operating room (sign out). In each phase, a checklist coordinator must confirm that the surgical team has completed the listed tasks before it proceeds with the operation. The checklist has been shown to reduce both patient morbidity and mortality in both developed and developing nations.58 There is now a drive for widespread use of the checklist.
The checklist is a very simple but effective technology that aims to enhance patient safety; however, other technologies that are already in use might be more difficult to redesign. An example of a piece of ongoing work at Imperial College is the redesign of the resuscitation trolley. Traditionally, this is no more than a workman's tool trolley, although absolutely crucial during a cardiac arrest. In the pressurised, time-critical and often crowded environment of a cardiopulmonary arrest, it has been shown that division of team roles with leadership and direction of resuscitation algorithms are often lacking. This is compounded by inaccessible contents and inadequate daily stock checking.59 Collaborative work between clinicians, nurses, psychologists, human factor specialists and engineers, using footage of real and simulated arrests, has led to a drawer-free open-layout resuscitation station with logical separation of equipment (for airway, breathing and circulation); radiofrequency identity technology has also been employed for instantaneous stock checking.60 The trolley incorporates an interactive touch screen to prompt the team leader and encourage appropriate role adoption within the team, while the software provides data capture for subsequent audit. Redesign and renewal is not always a financial possibility, however; simply understanding how errors are created is sufficient to change the practice of the end user. For example, heuristic violation assessment can be performed on widely used technologies such as infusion pumps to identify potential usability problems.61
The standard process of blood transfusion is an inherently dangerous process prone to human error. Through systematisation and multiple verification steps, much of the error from incorrect blood component transfusion has been removed. Bar coding to verify correct identification at multiple steps in the transfusion, such as a process to match patient and blood product at the bedside, has been introduced at the John Radcliffe Hospital in Oxford, UK.62 Hand-held computers are used to scan bar codes on the patient's wrist band and the blood product to ensure a match. Early data suggest that use of this system increases checking behaviours; long-term study will establish if there is a reduction in harm to patients.
Similarly, patient wrist band bar codes can be scanned together with medication bar codes to try to avoid human error.63 This increased complexity can have drawbacks,64 for many reasons including time constraints and the possibility of staff employing workaround strategies.65 However, human factor analysis within the sphere of intravenous drug errors in anaesthesia has demonstrated that the solutions need not be complex: there is evidence that prefilled syringes, colour-coding and syringe labelling immediately after drawing up the drug, structured organisation of drug drawers and different packaging and presentation of drugs in different classes help.66
It is imperative that all end users appreciate that intelligent redesign and safety systems will never eliminate risks from human factors. Indeed, measures taken to address human factors can increase complexity and, therefore, the propensity for errors due to technical failure. For example, the anaesthetic machine safety pins described in box 3 can be found to be missing or become worn and bent. Therefore, qualified and competent user vigilance is still required, along with continuous professional development to keep abreast of changes in technologies.
Box 3 How technology can be used to prevent human error within anaesthesia
Anaesthetic misconnections
While human error is responsible for most adverse incidents within anaesthesia, equipment inadequacies have been highlighted by the seminal papers on critical incident analysis by Beecher in 195467 and Cooper in 1978.68 In the 1970s, escalating litigation costs resulting from critical errors within anaesthesia catalysed critical incident analysis in a manner that had previously been practised in the airline industry. This analysis of near-misses and actual incidents led to developments in technology that provide early warning of, or prevent, human and equipment error. There was acceptance of national (eg, British Standards Institution) and international standards (International Organisation for Standardisation) for the components of anaesthetic machines. Safety advances include non-interchangeable screw threads between pipeline gases and the inlet on the anaesthetic machine, along with alarms and mechanical devices to prevent the delivery of hypoxic gas mixtures to the patient. Additionally, the pin index safety system offers protection against accidental connection of a pressurised gas cylinder to the wrong yolk. Each air, oxygen, carbon dioxide, heliox and nitrous oxide cylinder top has a unique arrangement of holes into which only the corresponding gas yolk's projecting pins can be inserted.
Key issues for the developing world
Insufficient attention to patient safety in low-resource, developing-world settings has been the result of a lack of awareness and inadequate financial, human and communication resources. In these settings, access to service and supplies is often limited, and there are major infrastructure gaps, such as outdated facilities, overcrowding, inadequate clean water, power and sanitation. There has been a perceived limited market for appropriate health technologies, and sometimes the only available technologies are designed for industrialised markets and are inappropriately complex. This causes both operating difficulty, with inadequately trained healthcare professionals of low literacy, and maintainance difficulties, particularly with extremes of temperature and humidity.69 Governance within developing-world healthcare is less advanced, and patients less aware of the risks and their rights; there is a power imbalance between patient and healthcare provider with poor reporting structures and legal recourse. Some argue that the increased presence and voice of professional medical bodies with their evidence-based guidelines within the developed world puts pressure on healthcare providers to provide safe technology for fear of institutional negligence. Within the developing world, fewer reports of concern and adverse incidents lead to continuation of poor practices. Standards and regulations to ensure product quality and safety can be inadequate, as well as the mechanisms to enforce them. As a marker of inadequate access to quality care, a patient in a low-resource health setting is at a 2–20-fold higher risk of acquiring a facility or Healthcare-Acquired Infection (HAI) than a patient in a high-resource setting, where approximately 5–10% of hospital patients suffer HAIs.70
Solutions need to be inexpensive to implement and designed for low-resource settings (box 4). For example, many developing countries have significant problems providing skilled attendance for obstetric emergencies. WHO Regional Office for Africa recommmends groups of young volunteers from the primary healthcare level transporting appropriate patients to referral level care using motorcyles or adapted vans, fitted with radio transmitters.74 Additionally, while it is sometimes culturally difficult, there is a limited evidence base for the provision of small and simple ‘maternity waiting homes,’75 situated close to the referral hospital. These homes are used by women in the final period of their pregnancy who are at risk of complications or by those who live far from the referral hospital.76 Where infants are born at home, infection risks are higher; however, single-use delivery kits provide a sterile plastic delivery sheet, razor blade, cord ties and soap with pictorial instructions for low-literacy users. Tanzanian studies have shown this reduces umbilical cord infection from 3.9% to 0.3% and puerperal sepsis from 3.6% to 1.1%.77 Another issue with home births by low-literacy traditional birth attendants is that detection of low-birthweight babies requiring treatment is more difficult. One novel solution has been the use of non-numeric tactile or colour-coded indicator weighing scales.78 Table 2 outlines some of the other challenges facing the developing world with relevant implications and potential solutions where possible.
Box 4 Development of auto-disable hypodermic syringes
Points of safety
Disposable plastic hypodermic syringes were developed in the 1950s, partly solving the problems caused by inadequate sterilisation of reusable syringes and needles. However, inappropriate reuse remains an issue, particularly within the developing world where cost and the reluctance towards single use are important factors. Reuse of syringes can cause transmission of nosocomial bloodborne infections, including HIV and hepatitis B and C, and has threatened the acceptability of immunisation programmes in the developing world. This prompted a 1986 WHO request for auto-disable syringe ideas. Over 400 designs were submitted, involving ideas such as immobilisation of the plunger, blockage of the needle and leakage when a second injection is attempted. Currently available auto-disable systems or syringes with reuse prevention features include the SoloShot, which has an internal metal clip to lock the plunger after injection to prevent refilling; the K1 syringe, which can serve curative injection needs as well as immunisation; and the Uniject, which is a prefilled single-dose non-reusable plastic bubble.71 All these systems can have integrated needles to prevent needle reuse and are now manufactured in more than 10 developing countries, priced at US$0.1–0.3 more than standard disposable syringes.72 Uptake has been boosted by Unicef replacing standard disposable syringes for vaccination programmes with autodisable syringes. Safety issues remain concerning the disposal problems inherent with any disposable needle,73 and a lack of needle protection for the prevention of sharps injuries. However, new low-cost needle protection systems are under development. Significant reuse is still prevalent in the curative sector, even within public health facilities. Reuse will hopefully become unnecessary as the market becomes saturated with K1 and similar devices that can replace all syringe sizes and types used in most procedures.
Clinical technological problems more marked in the developing world with implications and possible solutions
Conclusion
Although technology is pivotal for the advancement of healthcare, it can cause significant harm if not adequately designed, regulated and maintained. Although medical devices will have CE markings under the medical devices directive (93/42EEC) within Europe,92 and FDA approval in the USA, this level of certification must be ensured internationally and enforced within the developing world, especially for higher-risk technologies. It is likely that this would be easier to achieve with worldwide agreement on the minimum standards. Although it has proven difficult to acquire agreement on even common nomenclature, the Global Harmonisation Task Force has embarked upon this ambitious but important strategy.93 94 A recent example is the work performed by WHO and collaborators through the Global Pulse Oximetry Project to establish global standards for the use of pulse oximetry in anaesthesia.91
Even with robust regulation of minimal requirements for their design, however, healthcare technologies are vulnerable to misuse and can create error in ways that can only be identified through appropriate encouragement of non-punitive open reporting. By classifying and investigating these near misses and adverse events and recording them in national and international databases, it becomes possible to establish root causes. As described, data support the benefits of these systems with regards to safety of technology26 and they must be encouraged in the developing world.
It is time to start work on internationally standardised practical methodologies and nomenclature for reporting systems and on gathering information from all available sources based on the ICPS, which is scheduled to be completed in the next 3 years. A common data field format and means of collection will allow the development of a universal international events database. This is possible with limited technology such as mobile phones that are linked to reporting centres provided with the software that elicits the information needed for populating the ICPS. This data collection will pave the way for the final step—the dissemination and implementation of the lessons learnt. The cost of implementing basic systems for safe technology must be weighed against the current very high costs of not doing so.
Vital for the provision of safe technology are maintenance programmes and consideration of intelligent redesign to reduce the risks that are contributed by the end user. Furthermore, there needs to be adequate training and education programmes for healthcare professionals. Common standards for accreditation and quality assurance schemes will also improve safety. In the developing world, it is essential that all these safety mechanisms and solutions be affordable, appropriate and, above all, able to be realised.
References
Footnotes
Funding The project was funded by WHO Patient Safety.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.