Article Text

Statistics from Altmetric.com
Introduction
The US Cystic Fibrosis Foundation (CFF) began in 1955 with a mission to support the development of new drugs to fight the disease, improve the quality of life for those with cystic fibrosis (CF), and ultimately to find a cure for this disease.1 The CFF does this by supporting basic science and clinical research in CF, supporting the care of CF patients through accredited CF centres nationwide and advocating for CF patients at the state and national level.
Recognising the critical role of data collection and measurement of outcomes to better understand the natural history of CF, the CFF created a patient registry in 1966, the CFF Patient Registry (CFFPR).2 The CFFPR has evolved over the years from a few demographic variables including vital status to a comprehensive database that gives healthcare providers, researchers, policy makers and change agents data to support epidemiological and clinical research as well as efforts to improve quality of care.
The specific purpose of this commentary is to describe the CFFPR and primarily to focus on how the CFFPR and its associated tools are being used for quality improvement (QI) activities, with the hope that it may help CF healthcare teams in the USA who are not familiar with the registry's capabilities, CF providers outside the USA with registries at various stages of development, and others interested in how a patient registry has been used to improve care.
Description of the CFFPR
The CFFPR contains detailed demographic and diagnostic data dating back to 1986 with current annual and encounter-based data on over 300 unique variables including outcomes (eg, microbiology, lung function and nutritional metrics, CF complications) and care processes (eg, hospitalisations, medications, surveillance measures) for each of its more than 27 000 participants in 2012; in all, there are over 46 000 unique individuals’ data in the registry.3 Data are entered into a secure web-based portal (PortCF), with patient consent, by administrative and clinical personnel at CFF accredited care centres for whom training is made available at the annual North American CF conference and by means of a CFFPR Data Manual.
Significant resources are provided by the CFF to maintain the CFFPR, including 4–5 FTEs to provide user support, maintain documentation, conduct quality control measures on software upgrades, manage the change control process, design and manage the registry data warehouse, conduct data quality assessments and develop annual reports. When PortCF underwent a change in vendor and an extensive redesign in 2010, the CFF supported the development costs for the platform and all customisations and activation fees. To maintain the registry, the CFF pays for monthly hosting fees and project management support along with the fees for all enhancements to the registry platform.
Timely and accurate completion of data entry by CF centres is incentivised by the provision of monetary payments from the CFF that are proportional to the number and completeness of records entered into PortCF, as well as the CF team's ability to access and use the data for clinical care and QI work. Data reliability is also supported by automated checks at the time of data entry for in-range values. Manual audits of key variables have recently been initiated as an additional quality check, and will be the subject of a future report.
Evolution of the CFFPR and its contributions to knowledge of the natural history of CF
The initial stated goal of the CFFPR was to describe the CF population in the USA and track survival at a time when controversy existed around the optimal treatment of CF. Early reports of improved patient mortality at centres using a comprehensive treatment programme facilitated the spread of this multidisciplinary and proactive approach.4 This basic descriptive function, the documentation of the distribution of basic demographic and disease characteristics such as age, gender, rates of infection and comorbidities, in addition to outcomes such as lung function, nutritional status and survival, continues to give the CF community a broad perspective of the disease as well as ideas for new opportunities to improve care for specific subgroups of patients with CF.5 An example of this type of descriptive data is shown in figure 1, showing the cross-sectional relationship of lung function measurements with age in three separate historical cohorts.3 This kind of descriptive report represents the most basic use of a patient registry, providing important insights into the characteristics of the overall population, and was the primary function served by the registry in its earliest years.
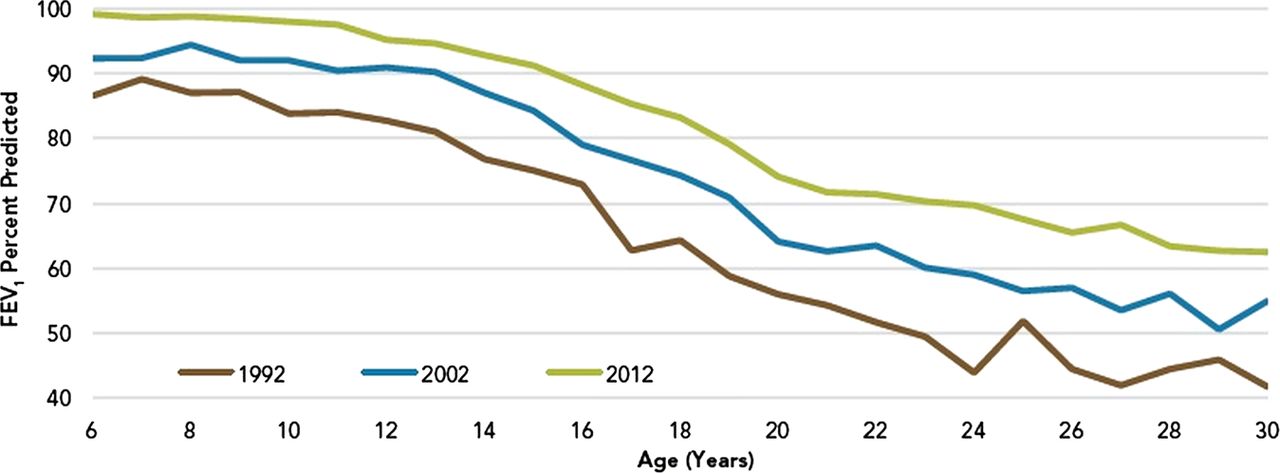
Median FEV1 per cent predicted by age for the cystic fibrosis (CF) population in 1992, 2002 and 2012.35 FEV1 decreases with age, but there have been notable improvements in this metric for the registry population over the last 20 years. This is an example of the initial basic function of the Cystic Fibrosis Foundation Patient Registry, which is to provide important descriptive information regarding the CF population to stakeholders in annual reports.
In 1995 the collection capabilities of the CFFPR were expanded and centres were asked to enter quarterly measures of growth and lung function as well as more detailed data on complications and treatment. Soon after, epidemiologists began to recognise that the CFFPR offered a resource to better understand the pathogenesis of CF and the opportunity to identify risk factors that may be associated with patient-level variations in disease course and outcomes. The majority of the 68 studies using CFFPR data published over the last 10 years have been risk factor analyses such as the impact on disease severity of gender, socioeconomic status, and acquisition of various microbial organisms in the airway.5–13
The use of the CFFPR registry as a driver for a national QI initiative
Under the influence of Gerry O'Connor, PhD DSc, a health services researcher who had previously worked with the Northern New England Cardiovascular Disease Study Group, the CFF began in the late 1990s to use the CFFPR to examine centre variations in practice and outcomes. Reports to CF care providers and eventually to the public began to showcase these variations, and centre directors began to receive private reports showing their centre’s position within the national distribution (examples are given in figure 2).3 The recognition of this variation led to serious self-examination by CF clinicians14 and was a vital stimulus to roll-out the CFF QI initiative (described in detail in another paper in this supplement).15 The availability and open discussion of CFFPR data at local and national meetings were needed for the culture change that led the CF community to embrace QI. Initial concerns that CFFPR data was not sufficiently reliable to allow appropriate and valid centre comparisons led care centre teams to work harder to ensure the accuracy of data entry. In addition, comparisons of outcomes are case-mix adjusted to account for differences in disease risk in populations attending different care centres; this is made possible by past CFFPR analyses exploring the impact of sociodemographic and disease-specific risk factors on disease outcome.11 ,16 ,17
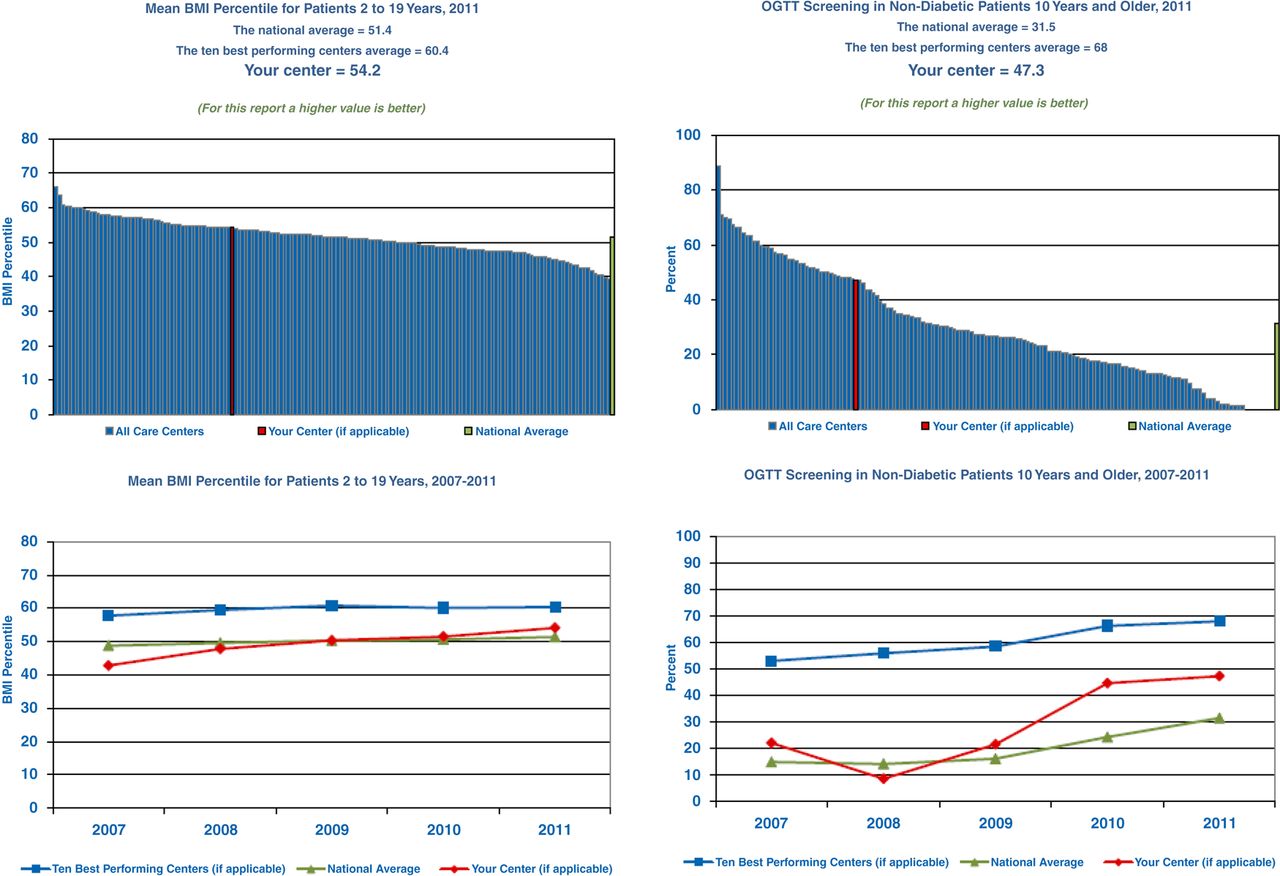
Examples of annual Cystic Fibrosis Foundation Patient Registry (CFFPR) reports provided to cystic fibrosis (CF) centre directors. The histogram on the left shows the distribution of an outcome: mean body mass index (BMI) percentile for patients 2–19 years of age in 2011. The vertical bars depict the average at each accredited CF care centre, with the care centre receiving the report highlighted and national average at the far right. The national average and that of the 10 best performing centres is provided, along with the average at the specific CF centre receiving the report. Below the histogram is a graph showing 5-year trends at the 10 best performing centres, the national average and the centre receiving the report. The histogram on the right shows the distribution of performance of a process: oral glucose tolerance test screening in non-diabetic patients 10 years and older, as recommended by CFF care guidelines.
Case-mix adjustment also set the groundwork for public transparency through reporting of CF centre outcomes at CF centre Family Education Days and on the CFF public website, CFF.org. The public reporting of outcomes was a crucial step undertaken by the CFF in 2006, affirming its commitment to patient-centred care and providing impetus to care centre teams to incorporate patient and family priorities into ongoing improvement work. A sample of the public report is shown in figure 3.

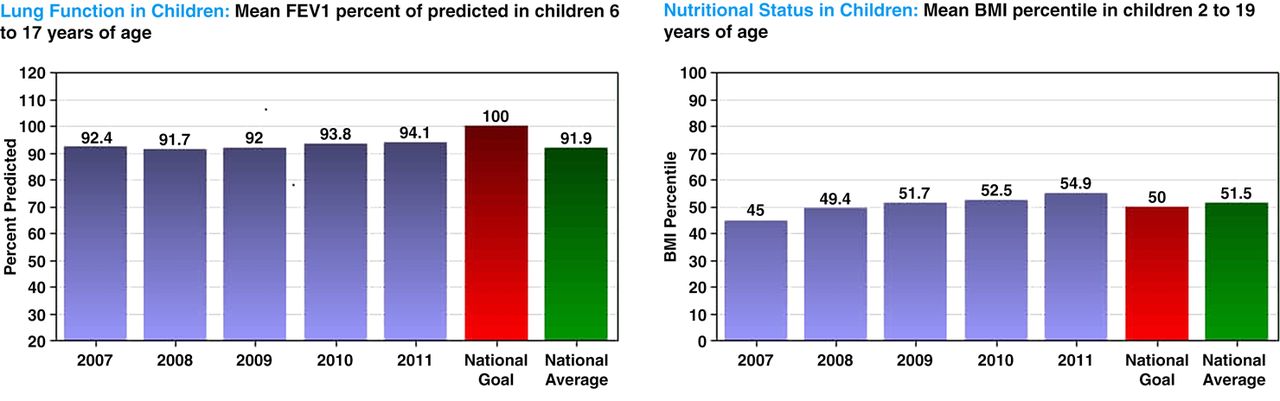
Programme-specific reports available to the general public at http://www.cff.org/LivingWithCF/CareCenterNetwork/CareCenterData. The figure on the left shows average lung function (FEV1 per cent predicted) in children by year over a period of 5 years and the current average of all care centres and the Cystic Fibrosis Foundation's (CFF's) declared goal; that on the right shows a nutritional outcome (BMI percentile). Other data displays available on the site are corresponding pulmonary and nutritional measures for adults followed at the care centre, and cystic fibrosis-related diabetes screening, and centre adherence to CFF guidelines regarding routine monitoring. Outcomes data is case-mix adjusted, and the webpage contains detailed explanations of the meaning of the data and a discussion of the margin of error of all estimates.
The ability to use the CFFPR to make reliable comparisons and ascertain care centres that attain the best patient outcomes also enabled efforts by the CFF to send out benchmarking teams in order to identify characteristics of the most successful CF care centres and the approaches to care that lead to their success. A description of that programme may be found in another paper in this supplement.18
Web-based data entry and the use of the CFFPR registry as a tool to sustain and support the national QI initiative
In 2003, the CFFPR transformed its data collection instrument from a paper-based year-end summary to an internet application called PortCF that provides a web-enabled, encounter-based format for data entry. Care centre teams can use Port CF to access current data in raw form or as preformatted reports to support both individual patient care and population-based management. The CFFPR data also provides the ability to develop QI goals, to track the ongoing effect of QI efforts, and to provide progress reports internally to care teams and externally to health system stakeholders and patients. This capability has facilitated presentations at the annual North American CF conference and in professional journals,19–23 including several of those included in this supplement. As a specific example, the efforts at Children's Healthcare of Atlanta (CHOA) to improve outcomes of hospitalisations for treatment of pulmonary exacerbations used regular downloads of CFFPR data to track recovery of forced expiratory volume in 1 s (FEV1) back to baseline,18 providing important feedback regarding the success of their efforts to the care teams.
Clinic end-user functions of the registry: PORTCF
Resources that end-users can access to support the goals of improving care and outcomes for CF patients include the following.
Individual patient reports
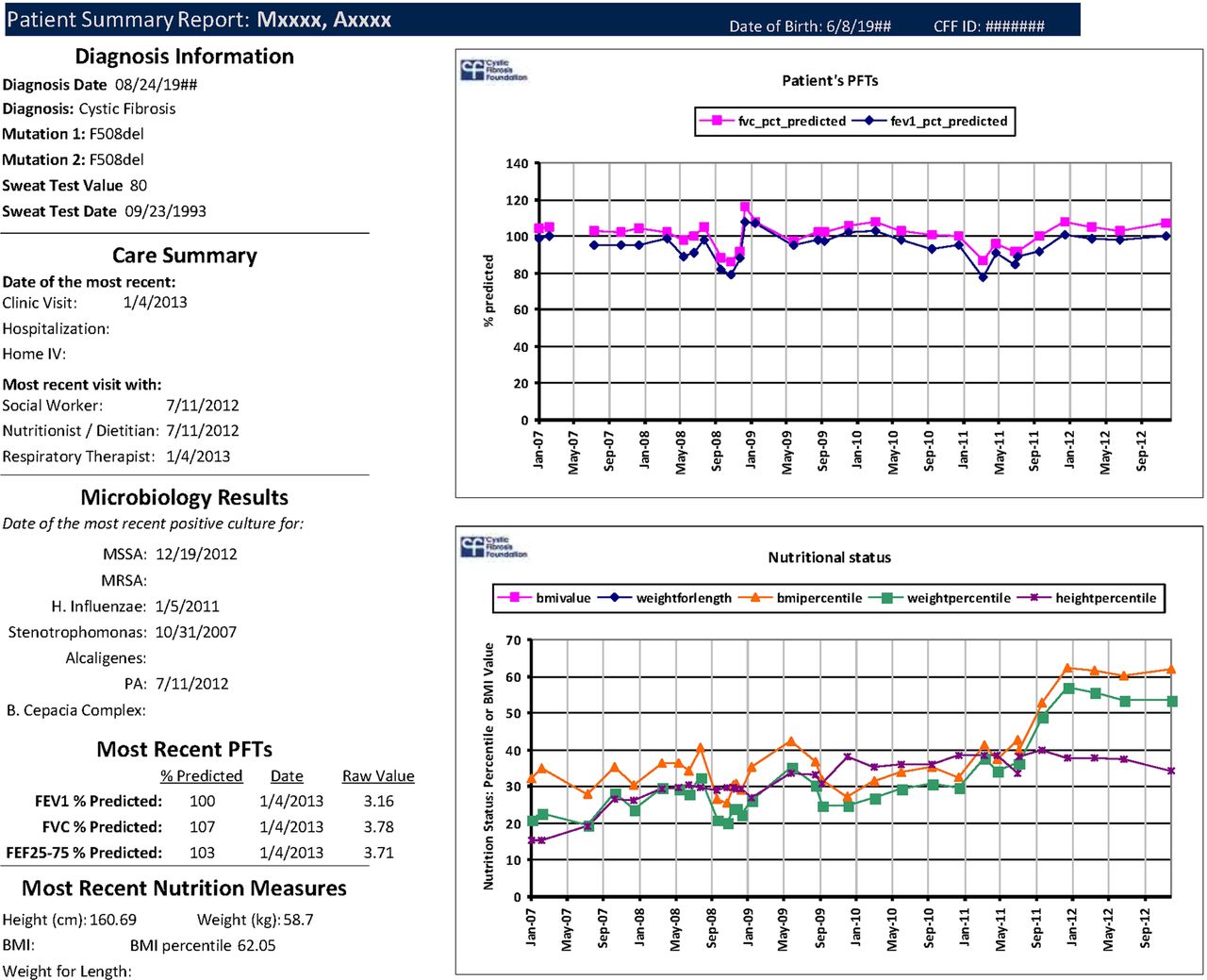
One of the most compelling benefits of the CFFPR to CF care providers is the value it brings during the clinical encounter for both patients and providers.24 CF centre staff can download patient reports from PortCF that provide structured data on clinic visits, hospitalisations, and longitudinal displays of microbiology, nutritional measures and lung function (figure 4). These reports are used by clinicians to prepare for patient visits at preclinic meetings, and may also be shared with patients to help initiate discussions to promote disease self-management. They are of particular benefit to care centres that are challenged by inadequate local institutional informational technology.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Example of a patient summary report downloaded from PortCF, based on data provided by the care centre. Note the longitudinal graphic display of lung function and growth and nutritional measures, as well as tabular representation of diagnostic data, visit dates and microbiology. Additional tracking of microbiology and other laboratory data is available as well. This report can be used for planning visits, and may also be shared with patients and families to facilitate disease self-management.
Centre level reports
Port CF has the capability to promptly and easily generate population-based reports and aggregate data. This allows the CF team to assess the consistency with which they are providing intended care and attaining target outcomes.24 They can generate histograms of key outcomes and processes (similar to those shown in figure 2) with up-to-date data they have entered into the CFFPR, providing real-time tracking of any process improvement efforts. Port CF contains tools to easily obtain a variety of preformatted reports, such as a list of patients who have not been seen in clinic in 3 months (the recommended interval) and 6 months; patients due for a nutrition or social work visit; patients who meet guideline recommendations for different therapies and whether they have been prescribed those therapies; and patients who meet entry criteria for various multicenter clinical trials being conducted through the CFF Therapeutic Development Network.
In addition to generating pre-formatted reports, PortCF also includes an application that enables the CF team to create custom queries. For more sophisticated data handling and analysis, care centres can download their dataset into data files that can be analysed using standard statistical software packages. These features allow unlimited customisation of data tracking and display so that the CF team may use the registry to follow any process and outcome measures reported to CFFPR for QI efforts. As an example, process control charts showing return of FEV1 to baseline following treatment of pulmonary exacerbations may be generated using this capability.18
Clinical tools
An additional section of the PortCF website, not directly related to the CFFPR, provides a searchable repository of over 800 documents including clinical tools, care guidelines, reports of all QI activities that have been conducted within the care centre network, patient educational materials, information about the CFF clinician mentoring programme and ways to access various CF listservs. Members of the care team can access this resource and avail themselves of a host of materials, much of it generated and shared by clinicians at other CF centres, that will help to enrich patient encounters and provide ideas to help generate new initiatives.
New and future directions and opportunities
The success of the CFFPR has led other countries around the world to develop their own CF Registries, leading to the possibility of international comparisons to be made as an extension of what are now predominantly US national benchmarking comparisons. A QI programme has been developed in Germany that uses their national registry to benchmark and then identify effective approaches to improve outcomes.25 The potential to discover novel approaches to optimal care is magnified considerably when the sharing of ideas occurs at an international level. An early example of this, with historically significant ramifications, was a comparison of nutritional outcomes and survival between the Toronto and Boston CF centres,26 which was instrumental in convincing CF care providers around the world of the benefits of a more aggressive and proactive approach to nutrition. Comparisons between registries is complicated by differences that exist in data collection procedures, which must be identified and reconciled in order to harmonise the data. Current work with Australia and the UK27 ,28 shows promise in allowing analysts to explore the effects of different care models and treatment approaches that are used in these countries.
With the recent and expected future expansion of availability of new medications to fight CF, there is an increasing desire among clinicians and researchers to use the CFFPR to document ‘real-world’ treatment effects,29–32 and for comparative effectiveness research. However, there are major potential pitfalls in attempting these analyses, most especially the problem of confounding by indication, whereby unmeasured indicators of disease severity influence treatment decisions; these threats to validity are an ongoing challenge to the use of any clinical registry, including the CFFPR, for this purpose.32–34 Nonetheless, early attempts to use the registry in this way appear to have been successful.32 In the coming years as an increasing number of therapies become available to treat CF, expectations are high that the CFFPR will be an important tool for determining the best treatment alternatives and combinations for clinical application.
Summary and implications
The CFFPR has played an essential role in the CFF efforts to improve quality of care and disease outcomes for patients with CF. The creation of provocative displays of centre-based variation served as an essential kick-start to the CFF QI programme and continues to provide ongoing motivation for engaged CF clinicians and staff to strive towards the delivery of better care; future international comparisons promise a potential sharing of new ideas. With PortCF, end-user care providers and patients are provided with clinical tools to help optimise the function of the care team and ensure consistent delivery of evidence- and consensus-based care. The CFFPR continues to fulfil its original intent, which was to describe basic characteristics and survival of CF patients, and has also evolved into an important research tool for epidemiologists interested in the effect of risk factors and individual variations in CF outcomes. Finally, it shows great promise for future investigations of comparative effectiveness of established and newly introduced CF treatments.
The experience of the CF community clearly demonstrates that the study and care of a rare disease can benefit from the availability of a robust disease registry. In recognition of this, CF registries have been established in a number of other countries and disease-specific registries have also been developed for tracking other rare conditions such as childhood cancers, complications of prematurity, inborn errors of metabolism, haemophilia and spina bifida.
Overall, the CFFPR has shown itself to be a vital resource for understanding the clinical course and optimal treatment of CF, an invaluable data support tool assisting clinicians to deliver reliable CF care, and essential component of the infrastructure for promoting systems-base improvement efforts across the CF network of care centres. Its singular success is attributable to the iterative efforts of the CFF to ensure that the CFFPR has evolved and grown in parallel with the needs and capabilities of stakeholders, and the organisation's commitment to provide resources to ensure its feasibility, validity and relevance for research and clinical needs and to establish its role and value in support of QI efforts. These are key lessons that should be considered by those who are planning the development of a disease specific registry for other conditions.
References
Footnotes
-
Contributors This article represents a synthesis of the ideas of all of the authors. The literature search was performed by MSS and CHG. The final version of the article was completed by MSS, but all of the authors contributed to its writing. MSS is the guarantor.
-
Competing interests MSS and CHG have received research support from the Cystic Fibrosis Foundation (CFF) MSS, KH, and CHG have served as paid consultants to the CFF CHG chairs the CFF Registry and Comparative Effective Research Committee, and MSS serves as a member.
-
Provenance and peer review Not commissioned; externally peer reviewed.