Article Text

Abstract
Background Postmarket surveillance of medical devices is reliant on physician reporting of adverse medical device events (AMDEs). Little is known about factors that influence whether and how physicians report AMDEs, an essential step in developing behaviour change interventions. This study explored factors that influence AMDE reporting.
Methods Qualitative interviews were conducted with physicians who differed by specialties that implant cardiovascular and orthopaedic devices prone to AMDEs, geography and years in practice. Participants were asked if and how they reported AMDEs, and the influencing factors. Themes were identified inductively using constant comparative technique, and reviewed and discussed by the research team on four occasions.
Results Twenty-two physicians of varying specialty, region, organisation and career stage perceived AMDE reporting as unnecessary, not possible or futile due to multiple factors. Physicians viewed AMDEs as an expected part of practice that they could manage by switching to different devices or developing work-around strategies for problematic devices. Physician beliefs and behaviour were reinforced by limited healthcare system capacity and industry responsiveness. The healthcare system lacked processes and infrastructure to detect, capture, share and act on information about AMDEs, and constrained device choice through purchasing contracts. The device industry did not respond to reports of AMDEs from physicians or improve their products based on such reports. As a result, participants said they used devices that were less than ideal for a given patient, leading to suboptimal patient outcomes.
Conclusions There may be little point in solely educating or incentivising individual physicians to report AMDEs unless environmental conditions are conducive to doing so. Future research should explore policies that govern AMDEs and investigate how to design and implement postmarket surveillance systems.
- Equipment and supplies
- medical devices
- physicians’ practice patterns
- influencing factors
- qualitative research
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- Equipment and supplies
- medical devices
- physicians’ practice patterns
- influencing factors
- qualitative research
Background
Medical devices are rapidly increasing in variety and complexity, and are a mainstay of patient care that sustain life and enhance health and quality of life.1 Medical devices include a wide range of health or medical instruments essential for the prevention, diagnosis, cure or treatment of a disease or abnormal physical condition.1 Those considered higher risk, or those associated with a greater likelihood of causing serious or adverse health consequences or death, include orthopaedic implants such as hip or knee joints, or cardiovascular implants such as pacemakers or implantable cardioverter defibrillators (ICDs).
Adverse medical device events (AMDEs), particularly those among higher risk devices with serious negative implications for patient outcomes, have garnered widespread attention through media reports and even litigation, and calls to action for implementing measures that balance access to innovative medical devices with strategies that minimise associated risk and enhance patient safety.2 Analysis of Food and Drug Administration (FDA) device malfunction reports from 1990 to 2002 found that the mean annual replacement rate was 20.7 per 1000 for ICDs and 4.6 per 1000 for pacemakers, and 61 deaths (31 ICDs, 30 pacemakers) were attributable to device malfunction.3 Among 30 002 devices approved by the FDA between 2005 and 2012, 249 were recalled, half during the first 2 years on the market.4 Registry data from Sweden and Australia showed that the 10-year risk of revision after primary total knee arthroplasty was 4%–6%.5 Among 70 orthopaedic devices approved between 1982 and 2014, there were 765 total postmarket changes (median of 6.5 per device), including 12 recalls and 168 alterations to device design.6 While such retrospective studies can identify AMDEs, data are not sufficiently timely to mitigate or prevent AMDEs in additional patients.
AMDEs arise in the months and years following implantation in many patients. Postmarket surveillance is a useful strategy for collecting and analysing a sufficient volume of data over time to more quickly identify devices prone to adverse events.1 Such data could alert industry to the need for device improvements, and alert device users to either avoid those devices in future patients or to use caution when implanting those devices, thus preventing potential future AMDES. However, registries do not exist in every jurisdiction or for every type of medical device. In the absence of systematic data collection, the identification and sharing of information about AMDEs rely on voluntary reporting by physicians, who are exposed to device-related problems first-hand and could potentially observe trends in their use of a device and its outcomes. Qualitative research in the UK, Australia and Canada suggests that individual healthcare professional factors (fear of blame, belief that errors were inevitable and it was pointless to report them, avoidance of bureaucracy, time constraints, lack of knowledge about what to report and how, cultural norms) and organisational factors (inadequate feedback, lack of processes and reporting systems) influence the reporting of medical errors.7–9
However, few studies have examined whether and how physicians report AMDEs, which are caused by device design or function, and may account for 10% of patient safety incidents in hospitals.10 Polisena et al interviewed 16 clinicians in two tertiary care hospitals to explore factors that influenced AMDE reporting.11 Participants said that views about incident severity influenced reporting and, when devices malfunctioned, they most often responded by discontinuing their use of the product. The Polisena et al 11 study suggests that individual healthcare professional characteristics and behaviour may play a large role in reporting AMDEs. However, it involved clinicians from two sites only who may have shared similar views. Moreover, research on the reporting of medical errors in general suggests that organisational or system-level factors may influence whether physicians report incidents.7–9
Additional research among a broader sampling of physicians is needed to explore the role of the individual versus the environment in AMDE reporting. If individuals have autonomy over AMDE reporting, education, incentives or other physician-directed interventions could stimulate greater reporting. If, instead, reporting of AMDEs is beyond the control of individual physicians, other interventions at the organisational or system level may be required. Understanding the interplay of these determinants could generate more nuanced insight on whether and how AMDEs are addressed, information crucial to developing and implementing processes or interventions that prevent or mitigate AMDEs in the future. The primary purpose of this study was to explore factors that influence AMDE reporting among physicians who use higher risk implantable devices from multiple sites.
Methods
Approach
Given little prior empirical study of the factors influencing AMDE reporting, a qualitative research design was used to gain a thorough appreciation of influencing factors, which could serve as the basis for future research.12 More specifically, an approach called descriptive qualitative research was used.13 This method focuses on gathering straightforward accounts of experiences rather than testing or generating theory. The critical incident interviewing technique was also used to prompt for relevant detail by asking participants to describe a particularly notable AMDE experience.14 Rigour and transferability were optimised by sampling respondents with differing characteristics based on their use or exposure to various types of devices; exploring responses inductively for emerging ideas including deviant cases; demonstrating responses from an array of respondents by including an anonymous identification code with exemplary quotes; comparison of independent thematic coding across multiple individuals; and complying with the Consolidated Criteria for Reporting Qualitative Research.15 The University Health Network Research Ethics Board approved this study, and participants provided written informed consent prior to being interviewed.
Sampling and recruitment
Purposive sampling was used to recruit physicians with a range of characteristics that could influence views and experiences: specialties that use different types of implantable devices including cardiovascular (cardiac or vascular surgeons, interventional cardiologists) and orthopaedic devices (orthopaedic surgeons); geographical region (different provinces in Canada) and hospital type (community and academic for orthopaedic physicians; academic only for cardiovascular physicians), which could impose different policies concerning device use and AMDE reporting; and years in practice (self-reported as early, mid and late career), which could influence individual physician behaviour. In our experience of recruiting physicians for qualitative interviews, 5%–10% of invited individuals agree to participate; therefore, we oversampled. Contact information was identified in publicly available directories of provincial certification agencies and on the web sites of hospitals and universities. Invitations and consent forms were issued by regular or electronic mail. A reminder was sent to non-respondents at 2 and 4 weeks. A minimum recruitment target was set at 10 of each specialty who varied in non-mutually exclusive fashion according to other sampling characteristics for an aim of 20 participants. However, as is common in qualitative research, sampling was concurrent with data collection and analysis and proceeded until no further unique themes emerged from successive interviews (saturation).12 Saturation became apparent prior to reaching the target of 20 interviews; however, we proceeded with additional interviews to balance the number of participants across orthopaedic and cardiovascular physicians and ensure that potentially differing themes were fully explored.
Data collection
Interviews of an average of 30 min were conducted via telephone by the principal investigator using a semistructured interview guide (online supplementary table S1). Participants were asked to describe a recent or common AMDE, including the type of device, nature of the incident and its impact (findings in the process of being published). This primed the participants to discuss AMDE reporting. They were then asked whether and how they report AMDEs in any fashion to any organisation or system, and to describe factors that influenced AMDE reporting, including individual healthcare professional factors such as knowledge and information seeking practices, and organisational or environmental factors such as policies or procedures in their department, hospital or region, or relationship with representatives of the medical device industry. Interviews were conducted between 8 April and 28 September 2015, audiorecorded and transcribed.
Supplementary file 1
Data analysis
The principal investigator, a PhD-trained investigator with expertise in implementation science and extensive experience in the use of qualitative methods to explore factors that influence patient and provider behaviours, identified unique themes in an inductive manner through iterative stages.12 Typical office software (Microsoft Word and Excel) was used to organise data. First, interview transcripts were read to identify, define and organise themes in participant responses relevant to each of the main interview questions (first-level coding). Second, a codebook was developed to organise codes reflecting emerging themes, their definition and sample quotes illustrating application of that code. Third, transcripts were rereviewed (constant comparative technique) to assess whether and how to expand or merge themes (second-level coding). Apart from the principal investigator, the research team was composed of eight investigators; five were investigators with expertise in social sciences, quality improvement, patient safety, human factors research, and health technology design, assessment and use, and three were clinician investigators with experience in surgery, implantable devices and patient safety. First-level and second-level coding were independently reviewed and refined by all members of the research team on three occasions, and saturation was determined by discussion and consensus among the research team on the third occasion. Data (quotes labelled by theme) were tabulated by theme and summarised. An anonymised participant identifier noted cardiovascular or orthopaedic device experience, geographical region and self-reported career stage. The summary was reviewed and discussed by the research team at a 1-day meeting to interpret data.
Results
Participants
Of 561 physicians invited to participate, 534 did not respond. Of 27 who consented, 5 were unresponsive to communication about scheduling an interview, and interviews were conducted with 22 (table 1). These included 8, 10 and 4 early, mid and late career physicians, respectively, from five different provinces. Of these, 10 interviews focused on cardiovascular and 12 on orthopaedic devices. Exemplar quotes are discussed here according to themes that emerged, with discrepant views or experiences noted where relevant.
Characteristics of interview participants
Multiple factors influenced AMDE reporting
Participants perceived AMDE reporting as unnecessary, not possible or futile. They described numerous factors at the individual, organisational, system and industry levels that all influenced AMDE reporting. Physicians were not motivated to report AMDEs because they viewed AMDEs as an expected part of practice that they themselves could manage by switching to different devices or by developing work-around strategies to continue using problematic devices. AMDEs were not monitored or reported locally, nationally, internationally or to industry due to a paucity of healthcare system conditions and motivators to promote or support AMDE reporting, the need to fulfil purchasing contract obligations, and lack of industry feedback or device improvement in response to physician reports of AMDEs. These factors further reinforced and constrained individual physician beliefs and behaviour related to AMDE reporting. As a result, some physicians used devices that were less than ideal for a given patient or with which they were unfamiliar, potentially leading to poor patient outcomes. The interplay of factors is depicted in figure 1 and discussed here with exemplar quotes.
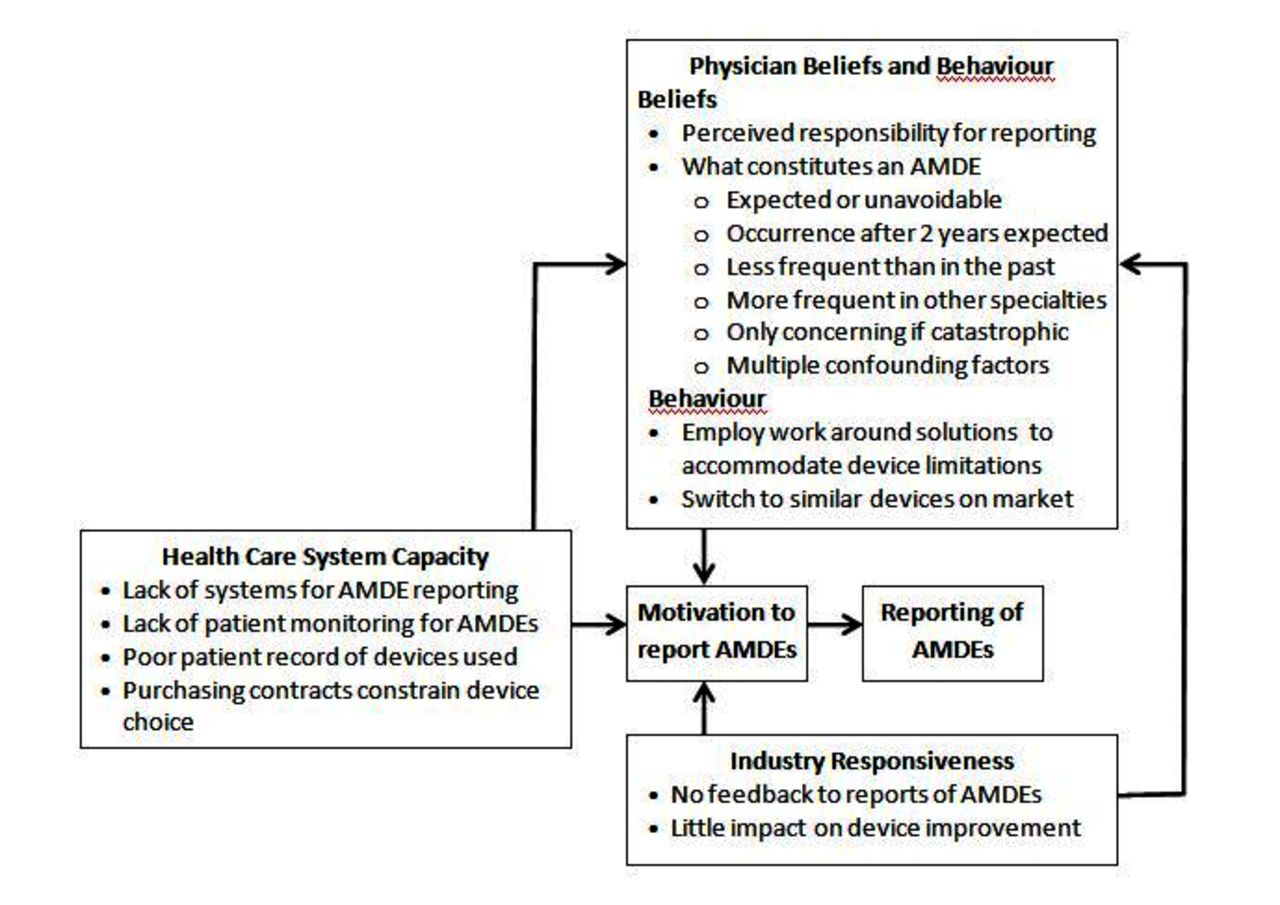
{kind=link}
Factors influencing adverse medical device event (AMDE) reporting.
Physician beliefs and behaviour
AMDE reporting was not perceived by participants as a need or responsibility. This was due to beliefs about what constitutes an AMDE, and because they could develop work-around solutions to accommodate AMDEs or avoid AMDE-prone devices by switching to comparable devices.
Perceived responsibility for reporting
AMDE reporting was not consciously perceived as a responsibility. Most participants said that they did not report AMDEs in any manner to the hospital, national or international organisations or systems, or industry. While most participants said they informally discussed device issues with colleagues, only one participant said that individual physicians should broadly share information about AMDEs by publishing that information.
“You could publish the results because, if you don’t do it, other doctors could use the device not knowing what happened. The whole field of medical devices is in the process of learning so every experience needs to be shared.” (18 cardiovascular, teaching hospital, late career)
Beliefs about what constitutes an AMDE
Participants said that individual beliefs about the probability, timing, incidence, outcome and cause of AMDEs influenced reporting. All participants thought that AMDEs were an expected aspect of medical practice. From the perspective of some cardiovascular surgeons or interventional cardiologists, events were not necessarily considered adverse because implantation of a medical device was the only life-saving option available to patients, and therefore unavoidable.
“Considering that these patients are facing life or death situations and the device is really their only chance at life it doesn’t really affect our use of the device.” (05 cardiovascular, teaching hospital, mid-career)
The timing of events also influenced whether they were perceived as adverse, or what would be considered normal device deterioration. This was more apparent among orthopaedic surgeons, who considered events as adverse if they occurred within a 2-year window.
“That locking bolt failed fairly early, within two years, which is very early.” (10 orthopaedic, community hospital, early career)
Several participants said that the incidence of adverse events had decreased over the years as devices were phased out or redesigned. This may suggest that, comparatively speaking, the lower incidence of adverse events was considered more acceptable. Perhaps because incidents were considered expected or unavoidable, only the most catastrophic of incidents were considered adverse. AMDEs were further downplayed based on the perspective that they were more serious or frequent in other specialties. Participants of one specialty assumed that physicians of the other specialty experienced a greater number or more serious adverse events.
Participants also said it was difficult to distinguish incidents directly caused by devices because multiple, potentially interacting factors were thought to influence AMDEs. Thus teasing out whether device failure contributed to AMDEs was challenging.
“It’s hard to know if it’s the device itself, the way the operator used it, or the way the patient’s anatomy might have changed over time.” (19 cardiovascular, teaching hospital, mid-career)
Behaviour in response to AMDEs
Instead of reporting AMDEs, participants either continued to use devices but developed their own techniques to manage AMDEs, or they discontinued use of devices that were prone to AMDEs.
Participants said they often developed a ‘work around’ solution to compensate for device limitations. The desire to continue using a given device despite the potential for AMDEs was said to be driven by physician comfort in using a product that was familiar to them and in which they had developed competence.
“The device actually had some great characteristics, in some ways it was very well designed, this was probably its major flaw [referring to multiple occurrences of nail misalignment]. So there was a kind of work around to get around the limitation of the device.” (06 orthopaedic, teaching hospital, mid-career)
In contrast, a few participants said that competence in only one device was not necessarily in the best interest of the patient. Thus, if one device performed poorly, physicians open to trying new products could choose from an array of similar devices on the market. Thus there was little need to dwell on or report problems inherent in a given device given that they had other similar devices to choose from.
“Some people may feel they’re only comfortable sticking with one. But being an arthroplastic surgeon is complex. You need a variety, and there are benefits and down sides to every single implant in terms of correcting for deformities, problems, variations on normal anatomy. In order to give the best outcome for patients, the one or two implants that you’re comfortable with may not correct those issues and that’s why I feel the need to use a wide variety of implants.” (14 orthopaedic, teaching hospital, early career)
Healthcare system capacity
Participants noted an absence of environmental conditions conducive to AMDE reporting. They said that incentives, processes and infrastructure were lacking in their own hospital and more broadly by which to detect, capture, share and act on information about AMDEs.
Lack of systems for AMDE reporting
Participants described an absence of local, national or international policies, processes or databases to promote and support AMDE reporting. A few cardiovascular physicians said that they contributed to international registries for specific devices.
“I’m not sure that there actually is a process to be very honest.” (21 cardiovascular, teaching hospital, mid-career)
“We do have what are called critical incident reports but that covers all aspects, it doesn’t necessarily look at devices.” (09 orthopaedic, community hospital, late career)
Lack of patient monitoring to identify AMDEs
Most participants said that they did not monitor adverse outcomes among individual patients in whom devices were implanted beyond the recovery period. AMDEs, which may arise in the months and years following device implantation, could be missed given the lack of active surveillance by the implanting physician, and the uncertainty of subsequent surveillance or AMDE identification by other physicians.
“The only real monitoring that goes on is we put something in, we follow it up to see whether it healed and the problem went away.” (12 orthopaedic community mid-career)
Poor documentation of devices in patient records
Participants said that patient medical records lacked details about devices that were implanted. Thus, if a particular device were recalled, it would be difficult to identify patients who had received the device in question in order to alert them or replace the device.
“It was actually a whole lot of leg work on our part. The company sends out the list of batch numbers that were affected and we had to go through all of our patient records to find out whether anybody had it. We have a stack of notebooks with patient information and stickers from the product so we had to keep going through it and looking for the lot numbers back through the period of time the company says that the lot number was produced.” (12 orthopaedic, community hospital, mid-career)
Purchasing contracts constrain device choice
Participants said that purchasing group contracts between hospitals or health regions and manufacturers or distributors required them to continue using specific devices even if the device was not the most suitable or resulted in suboptimal patient outcomes. Thus, even if a device were prone to AMDEs, physicians practising under purchasing contracts may not be able to switch to another comparable device available on the market and, given these constraints, would not be likely to report AMDEs.
“The buying group…pushed down the price of implants from the manufacturer because we buy in bulk…I find that a bit disturbing but apparently it’s cheaper for the hospital…and sometimes the implant you put in is not what you think is the best for the patient because that’s the only thing available through the buying group.” (07 orthopaedic, teaching hospital, late career)
“We’ve had experience that if you force surgeons to change implants based on a contract that your complication rate goes up for a while. So it makes good business sense until you actually go and look at your revision costs over the next months to two years and then, all of a sudden, all of your cost-savings went into pain and suffering of patients and their subsequent care.” (08 orthopaedic teaching hospital, mid-career)
Industry responsiveness
Lack of industry responsiveness to reports of AMDEs was said to further limit AMDE reporting.
No feedback to AMDE reports
When participants reported AMDEs to industry representatives, they received no feedback about whether or how that information was processed by industry representatives, distributors or manufacturers, which in turn may have reinforced physician beliefs and behaviour such that they did not feel it was their responsibility to report AMDEs and they should continue to develop work-around solutions or, in the absence of contractual obligations, to use different devices.
“I just submit my complaints to them. What they do with it, I have no idea.” (20 cardiovascular, teaching hospital, mid-career)
Little impact of AMDE reporting on devices
Participants suggested that manufacturers were not likely to use reports about AMDEs. They said that AMDEs might be addressed by industry if they were likely to influences sales, but not necessarily in a timely manner. This likely further reinforced the perceived futility of AMDE reporting.
“Eventually the manufacturer paid attention and then changed the engineering of the device. But that took probably somewhere between two and three years. They’ve invested a huge amount of money in product development and then they’ve got a big back inventory. So if there’s a big cost associated with change, and people have figured out a work around, then there’s a lot less pressure on the company to change.” (06 orthopaedic, teaching hospital, mid-career)
Discussion
This study was conducted to explore factors that influence whether and how physicians who use higher risk implantable devices report AMDEs. Multiple physician, healthcare system and device industry factors influenced individual physician views and behaviour such that they perceived AMDE reporting as unnecessary, not possible or futile. As a result, some physicians switched to using different devices while others continued to use problematic devices with which they had developed familiarity, or which they were obligated to use given purchasing group contracts, potentially leading to poor patient outcomes. Overall, the views and experiences expressed by participants were similar regardless of specialty, years in practice, type of hospital or geographical region.
Strengths of this study included purposive sampling to recruit participants who varied according to a number of characteristics, which may have influenced their views or experiences, including specialty, type of organisation, geographical setting and years in practice, and rigorous methods for data collection and analysis. More importantly, we sampled to thematic saturation, in other words to the point where no further unique information emerged from successive interviews, which, in qualitative research, signals that recruitment is sufficient. Still, the interpretation and application of these findings may be limited by several factors. The total number of participants may appear small to those not familiar with qualitative research. Qualitative research is meant to capture detailed information from participants with characteristics representative of a larger group or population of interest, or characteristics that vary and could potentially influence views and behaviour. Additional organisational factors that influence AMDE reporting may have been revealed if we had sampled physicians from hospitals with various characteristics, for example, single hospitals versus multihospital corporations, hospitals with and without electronic medical record systems, or hospitals with higher and lower performance ratings. Participants were sampled from Canadian hospitals so the findings may not be transferable to other settings. Our participants use the same types of devices, and described similar behaviour with respect to AMDE reporting and similar factors influencing that behaviour regardless of variable characteristics. Given that physicians elsewhere use the same devices, the AMDE reporting experiences reported here may be broadly relevant.
These findings confirm and expand on previous research by Polisena et al, 11 which identified physician factors that influenced AMDE reporting.15 Little other research has investigated AMDE reporting. For example, a Japanese study showed that, for 6610 cardiovascular AMDEs which included death in 9.2% of cases, 51% of AMDEs were not reported by industry to the Pharmaceuticals and Medical Devices Agency of Japan within 15 days, and 10.9% were not reported to healthcare providers within 15 or 30 days as is required by law.16 Some research has examined the use of automated algorithms embedded in electronic medical records to identify short-term adverse outcomes among patients in whom devices were implanted.17 Adverse events can also be detected by electronic sensors within medical devices that automatically submit data to physicians or a monitoring agency when irregularities arise, although these are in various stages of development and testing.18 Other research exploring the reporting of medical errors in general found that, similar to this study, individual factors (healthcare professional view that errors were inevitable and it was pointless to report them) and organisational factors (lack of reporting processes and systems) influenced reporting behaviour.7–9 However, other factors that emerged in those studies, for example, healthcare professional’s fear of blame, did not emerge in our study. Furthermore, factors unique to those that influenced the reporting of medical errors emerged in our study that are specific to AMDEs, for example, purchasing contracts and limited industry responsiveness that reinforced the under-reporting of AMDEs. Therefore, to the best of our knowledge, our findings are novel. In particular, no previous research empirically examined the confluence of individual and environmental (organisational, system, market) factors that influence AMDE reporting as a means of generating insight on postmarket surveillance, or documented the fact that the onus is on individual providers to recognise, report and resolve AMDEs as there is little local, national or international support or imperative for them to do so.
Reporting might be enhanced if physicians were more aware of what constitutes an AMDE that warrants reporting. Audit and feedback is a process that appeals to professionalism by providing individuals with data on their performance to stimulate behaviour change, and has been used successfully in other contexts.19 With respect to AMDEs, feedback could be used to provide physicians with general data on the type, frequency and outcomes of AMDEs, or physician-specific or department-specific data on adverse events and their impact. Another strategy for creating broad awareness of when and what to report would be to provide guidance for AMDE reporting through informational or educational strategies. Other groups have developed guidelines for the reporting of adverse events in usual practice and in the context of clinical trials to achieve greater reporting consistency between clinicians, investigators and institutions.20 At this level there is a potential role for advocacy and stewardship by professional societies.
However, there is little point in priming physicians to report AMDEs if there are few or no environmental-level mechanisms for doing so, as was revealed by this study. Medical devices are essential to the treatment and well-being of patients, and the industry is driven by market forces. Therefore, it is unclear if an actionable solution is available at the manufacturer or distributor level. Others have noted that, due to the nature of device development and marketing, it is unlikely that the licensing criteria and process will change, and stronger premarket regulation will not prevent all device-related incidents since they may not occur or be recognised for several years.1 National and international registries are viewed as valuable resources but are costly to develop and maintain, and have thus only been feasible for a few, select devices or jurisdictions.21 Others have recommended multifaceted approaches. For example, an editorial suggested that a national surveillance system includes tracking with device identifiers; mandated reporting for high-risk devices to independent academic or professional organisations; financial contributions from industry; automated prospective analysis of registry data; and linkage between premarket and postmarket registries.2 A group representing industry, regulators, academia and professional societies suggested improving and enforcing regulations, linking premarket and postmarket strategies, creating financial incentives for participation in postmarket studies, encouraging postmortem device retrieval and aligning professional societies with national regulators.22 Clearly, postmarket surveillance of medical devices is a complex issue with no clear or easy solution for improving the reporting and sharing of information about device performance and associated outcomes.
Future research should evaluate whether AMDE reporting guidelines, audit and feedback or other strategies can promote AMDE reporting by physicians. However, given the absence of systems to capture and share this information, there may be a greater imperative for research that investigates how to design and implement postmarket surveillance systems. One avenue is to activate public reporting of AMDEs, perhaps through social media, as a supplement to regulatory agency reporting.23 However, the problem may well be beyond the scope of what research can achieve and may require greater attention at the policy level. Policy analyses have revealed a lack of direction for other types of health system innovations. For example, Wiig et al 24 found that health policy in 10 European countries did not specify mechanisms to improve healthcare quality. Gauld et al 25 found that primary care policies in seven countries only recently identified quality and safety as important platforms. Therefore, research on whether and how legislation and policies recognise, promote and incentivise AMDE reporting may be a useful first step in identifying an actionable gap that, if addressed, could contribute to improved postmarket surveillance of medical devices.
Conclusion
Participants perceived AMDE reporting as unnecessary, not possible or futile. Physicians were not motivated to report AMDEs because they viewed AMDEs as an expected or unavoidable part of practice that they themselves could manage by switching to different devices or by developing work-around strategies to continue using problematic devices. Device industry factors (no feedback to reports of AMDEs, little impact on device improvement) and healthcare system capacity (lack of systems for AMDE reporting, lack of patient monitoring for AMDEs, poor patient record of devices used, purchasing contracts constrain device choice) reinforced individual physician views and behaviour. As a result, some physicians used devices that were less than ideal for a given patient or with which they were unfamiliar, potentially leading to poor patient outcomes. Interventions are needed to promote and support AMDE reporting; however, there may be little point in educating or incentivising individual physicians to report AMDEs unless environmental conditions are conducive to doing so. Future research should explore policies that govern AMDEs and investigate how to design and implement postmarket surveillance systems.
Acknowledgments
The authors would like to thank Julie Takata for the assistance they received in organising meetings for this study.
References
Footnotes
Contributors ARG and DU conceptualised the study and acquired funding. All authors designed the study, collected and analysed data, drafted the manuscript, and gave final approval of the version to be published.
Funding This study was funded by the Canadian Institutes for Health Research.
Competing interests None declared.
Ethics approval University Health Network Research Ethics Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data are available in the article or online supplementary files.
Correction notice This article has been corrected since it published Online First. The author Patricia Trbovich’s name has been corrected.