Article Text

Abstract
Aim This report considers the introduction of new technology and the implications for patient safety.
Methods and discussion A distinction is made between ‘conceptually’ new and ‘contextually’ new technology. The life cycle of technology from development to routine use is discussed and the key role for regulation, health technology assessment, clinical engineering and surveillance in this life cycle considered. The limitations of each of these disciplines are also discussed. Special consideration is given to the needs of developing countries. Case study examples of particular challenges in the safe introduction of technology are presented.
- Patient safety
- devices
- appropriate technology
- regulation
- health technology assessment
- medical error
- risk management
- training
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Patient safety
- devices
- appropriate technology
- regulation
- health technology assessment
- medical error
- risk management
- training
Introduction
Technology has transformed medicine. Medicine practised today is virtually unrecognisable from that practised at the turn of the last century. We have new diagnostic aids, ranging from rapid tests for malaria to high-tech scanning equipment such as MRI. There are new preventive therapies, such as vaccination, and therapeutic interventions such as laparoscopic surgery. Technologies to support the delivery of medical care, such as telemedicine and the use of electronic medical records, have also advanced. Moreover, the internet puts tremendous power and knowledge in the hands of patients, as well as making knowledge readily accessible for many healthcare practitioners worldwide. Technology has also been instrumental in advances within patient safety, from the design of single-use auto-disable syringes to electronic prescribing.
New technology has the potential to improve the quality, efficiency and safety of healthcare delivery; however, introducing new technology can be a sensitive process in some healthcare settings and could introduce new risks and unforseen dangers. New technology can be conceptually new or contextually new. Conceptually new technology is novel or genuinely new technology that has only recently been developed or invented. For example, laparoscopic surgery in the 1990s was a conceptually new technology.1 The safety profile was unknown, and the potential uses were still in development. By contrast, contextually new technology is the introduction of an established technology to a new environment. The technology might be new to the staff and setting—for example, the introduction of angiography to a provincial hospital in a developing country that has no previous experience of angiography. The safety profile of this technology should be well established, although there is still a clear need for staff training and a requirement to learn about how the technology will function safely in that particular setting.
The Health Technology Resolution of the WHO World Health Assembly defines health technologies as ‘the application of organised knowledge and skills in the form of devices, medicines, vaccines, procedures and systems developed to solve a health problem and improve quality of lives.’2 Safety is defined as the ‘condition of being safe from undergoing or causing hurt, injury or loss,’3 whereas patient safety is concerned with preventing medical error that might lead to adverse events and harm to the patient.
Any judgement on safety must always take into account the severity and prevalence of the disease the technology is intended to treat. For example, if a disease is severe with a high risk of mortality, a technology might be judged as safe, even if using the technology involves a relatively high risk of morbidity or even mortality. The same level of morbidity or mortality would be completely unacceptable for less severe disease. Similarly, preventive technologies such as vaccines might be considered unsafe in an area of low disease burden, but might be considered safe in an area of high disease burden. Even with a highly effective life-saving technology, however, risk must be kept as low as possible.4
This paper focuses on the impact of new technology on patient safety, rather than on occupational or environmental safety. Emphasis is placed on technologies with an explicit clinical emphasis, both diagnostic and therapeutic, including devices, software, equipment and procedures.
Life cycle of technology
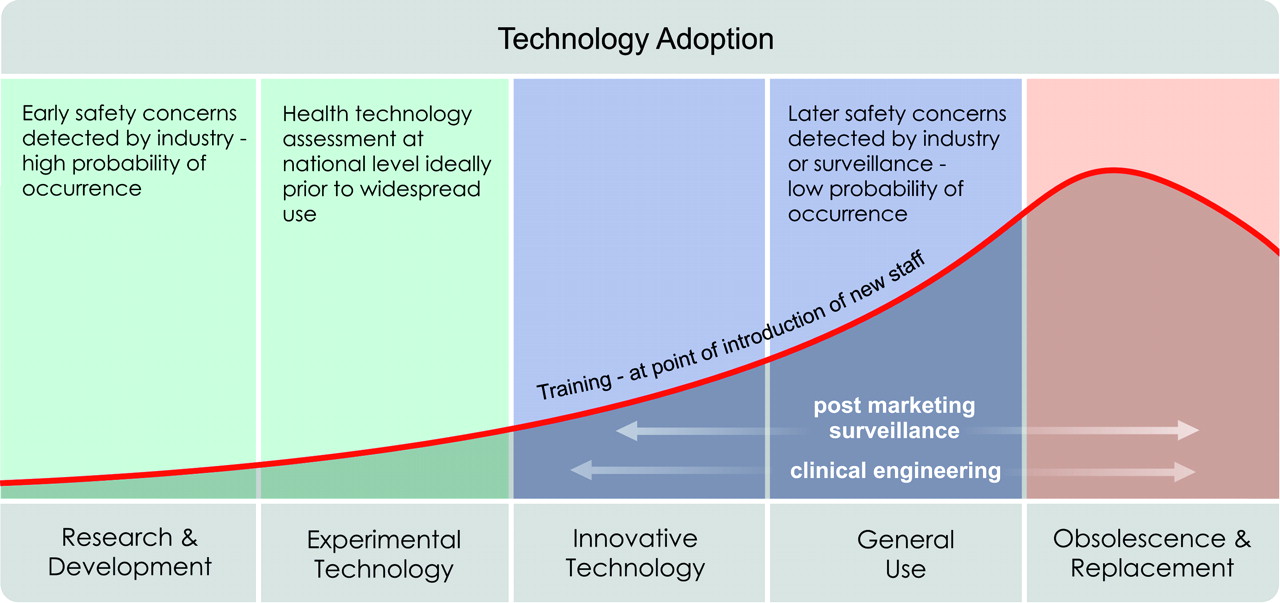
There are a series of stages during the development and introduction of technology, shown in figure 1. In an ideal world, a conceptually new technology is developed in consultation with end users. The new technology is then tested in a safe environment, perhaps using simulation, before having real-world trials that are regulated by an ethics committee. Evidence gathered during research, development and early clinical trials is key to informing decisions around the safety of a device before its widespread use. Decisions about the safety of a new technology can be made at a variety of different levels depending on what is required in relation to the end user (see box 1). Before marketing and widespread use, the technology will undergo a formal procedure for certifying that it is safe to be used.
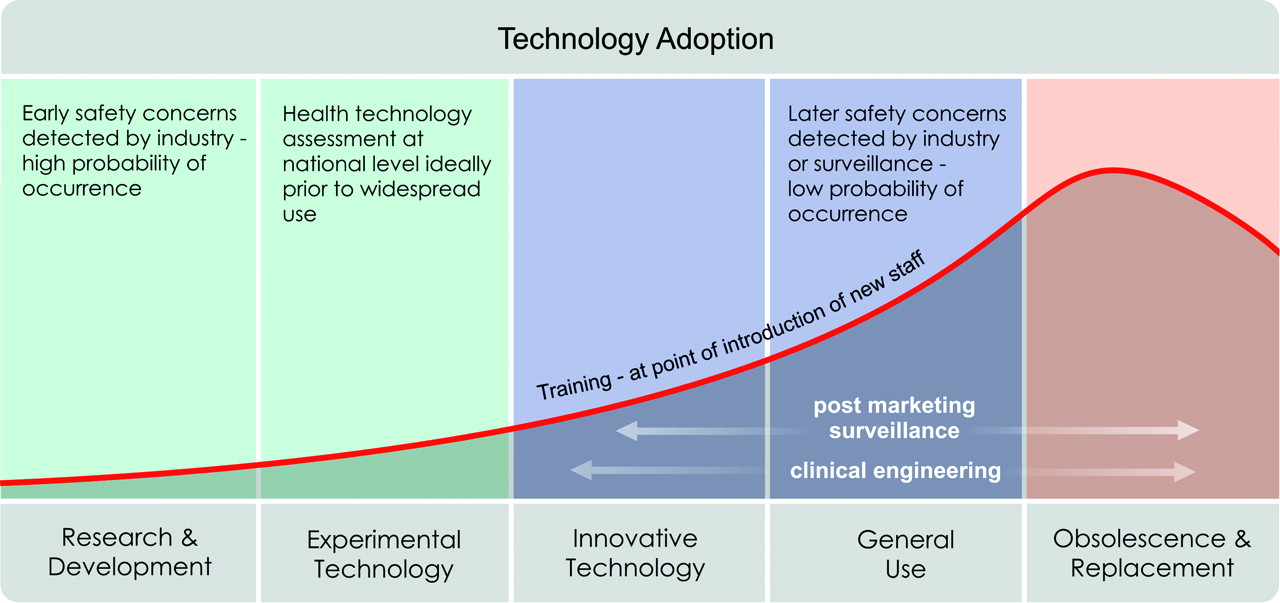
Life cycle of technology. Different safety concerns emerge through out the life of a new technology.
Box 1 Different levels of decision-making
Micro (either doctor or patient)—Is it safe to use this new device on me or this patient? Is it safe to use this device for a different procedure than it was originally intended?
Meso (hospitals, regions)—Issuing local guidance on safety and use. Do staff need retraining?
Macro (health policy)—The focus on effectiveness or cost-effectiveness with safety embedded into the concept of effectiveness. Is this device safe to be endorsed and used in this country?
In reality, the extent to which this happens is highly variable across the world. In Europe, before any medical device is marketed, it must have Conformité Européene approval. This indicates conformity with the essential health and safety requirements set out in the European Directive. For all but the lowest risk devices, such as a simple bandage, this approval is given by an independent certification body or a Notified Body; for low-risk devices, this is achieved by the manufacturer's internal self-certification process.5 A similar process operates in the USA overseen by the Food and Drug Administration (FDA). Unfortunately, on the whole, regulation is largely absent in many countries, and the means to introduce technology are not standardised. Health technology assessment is a separate process that is largely independent of regulatory decisions. Thus, only a minority of technologies are subject to a health technology assessment, even in developed countries.6
In this paper, the key stages in the development and introduction of a new technology are highlighted, including regulation, Health Technology Assessment (HTA) and clinical engineering. Aspects of training and surveillance are also considered.
Key stages in the introduction of new technology
Regulation
Regulation aims to ensure that technology performs safely and effectively. As no medical devices are risk-free, regulating medical devices is essentially a risk management process. It tends to consider whether a device performs when used correctly and under ideal circumstances, rather than in a real operating environment such as a busy hospital ward. To protect public health while ensuring continued access to new technologies and an innovation-friendly business environment, the level of regulatory controls should be commensurate with the potential risks associated with the device. The level of regulatory control should increase with increasing degree of risk, taking account of the benefits offered by use of the device. At the same time, the imposition of regulatory controls should not place an unnecessary burden on regulators or on trade and industry.
The regulatory framework should address the entire product life cycle of a medical device, from conception to disposal. It is recommended that the framework be drawn largely in line with the guidance documents recommended by the Global Harmonisation Task Force (http://www.ghtf.org), including definition and classification of the medical device, essential principles of safety and performance, quality system requirements, vigilance system requirements and the use of international standards. Moreover, the use of international nomenclature and standards is encouraged, as there is clearly potential for duplication and inefficiency with similar organisations repeating work. International drug names aim to standardise names across countries, avoiding mistakes when doctors work in different healthcare systems. It is also more efficient for look-a-like and sound-a-like names to be resolved internationally. Furthermore, countries should look for opportunities to cooperate in postmarket surveillance and incident-reporting activities to help set up an international medical device database that allows timely and global access to medical device safety information.
For all the benefits of global harmonisation, there can be no single template that responds to the needs of all countries. Some modifications must be made to suit local circumstances. However, the introduction of country-specific requirements will lead to inefficiencies, such as increases in the costs of medical devices and delays in approval. Such delays could hinder timely access to new healthcare technologies, with the result that the safety of patients can be unwittingly jeopardised. Countries must balance the need of local adaptations to the framework with the benefits they can reap from global harmonisation. If the globally harmonised model for regulating medical devices is adopted, consumers will benefit from internationally accepted best practice and timely access to new and safe devices.
Health Technology Assessment
Health Technology Assessment (HTA) is a systematic, transparent, unbiased and robust evaluation of a technology. The key component is a systematic literature review. The evaluation might include information about clinical effectiveness, social, economic, safety, organisational and ethical issues related to the use of technology.7 The HTA aims to inform the formulation of safe, effective, sustainable health policies that are patient-focused and seek to achieve best value;8 the assessment is rooted in research and scientific methods. If few or no data are available, for example where new technologies are concerned, grey literature or expert opinions can be included in the assessments. HTAs used to be produced for more complicated or contentious decisions, such as the use of bariatric surgery to treat obesity or which hip prosthesis to use.9 However, these assessments are increasingly used for decision-making at the local and regional level as well as nationally, as shown in figure 2.

Questions about safety. Different questions about safety are asked at different stages in the introduction of technology.
Safety is one of several parallel strands, alongside others concerning effectiveness, economic and ethical issues, involved in an HTA.10 Although not all HTAs explicitly assess safety, nearly all consider effectiveness or cost-effectiveness.11 Patient safety may be considered under headings related to effectiveness. If a device is causing patient harm such that it increases mortality or morbidity, it is unlikely to be considered acceptable for clinical use even if the technology itself proves to be ‘efficient.’ Moreover, harm often increases costs, which might become identified by economic or cost-effectiveness analyses.
It is also important to consider the way the new technology is causing harm—device failure, operator-dependent or setting-dependent. The type of harm should be noted and, if possible, quantified (eg, as disability-adjusted life years or quality-adjusted life years). Such quantification puts harms on a recognised scale of benefits making comparisons easier. Safety can be summarised as the frequency of adverse events (best displayed relative to a meaningful control group) or the number needed to harm. Relative risks are frequently used, but should not be the preferred means to convey safety risk to policy makers. Different sources of information, surveillance, observational studies and randomised controlled trials will have different strengths and weaknesses, and these should be acknowledged in any HTA.7 Patient and public participation is often needed to provide value judgements for balancing risks and benefits.12
Although the resources needed for producing extensive HTA reports are limited for many developing countries, HTA evidence produced elsewhere can be modified or adjusted for the local context. International organisations such as Health Technology Assessment International (http://www.htai.org/) and the International Network of Agencies for Health Technology Assessment (http://www.inahta.org/) can provide support to identify and use HTA reports produced elsewhere in the world. All countries should consider the introduction of receptors for HTA information into the different decision-making bodies in their health systems. HTAs should be used as a comprehensive assessment of safety, effectiveness, economic, social, ethical and organisational issues when complex new technologies are introduced in health systems.
Clinical engineering
Clinical engineering is concerned with selecting and implementing medical technology to ensure safe and effective operation. The focus is on the practical implementation of technology in the healthcare setting. Simply owning the necessary technology is not the complete solution.
Clinical engineers, who specialise in healthcare technology, need to participate in the planning, evaluation, selection and implementation process of both conceptually and contextually new technology. This might involve a direct contribution to local health technology assessments. Selection is a key step: having well-chosen equipment that matches the clinical need, as well as having a device that is easy to use yet accurate and reliable in the environment for which it was chosen, contributes to safe outcomes and effective healthcare. Equally important is the need for evaluation, to ensure that the intended benefits are realised and to identify harms. Careful planning is needed when a new technology is introduced. Decisions need to be made about how the technology will fit within an organisation's work, when staff will be trained and how the technology will affect existing services. Consideration should be given to the operating environments (eg, the quality and reliability of electrical power) as well as the skills and needs of the users.
Procurement of medical technologies might focus on the lowest-cost product, although the lowest cost products might not be the safest devices. Similarly, the reliability and availability of spare parts, ease of use, length of training required and safety record should be considered. Ultimately, devices that are difficult to maintain or have a poor safety record will cost more in the long term.
Some new technologies will require specialist training for staff so that they understand the correct indications and means to use the new technology. The type of training necessary will be determined by the technology, the risk it poses and the extent to which it is novel. For example, non-invasive surgical techniques offer a radically different way of operating in comparison with open surgery, and there is a clear need to train surgeons in this new technology. This recognition facilitated the development of virtual reality simulators for training surgeons. By contrast, a new blood-pressure-measuring device might require minimal or no training among existing staff.
The responsibility for training might be unclear. When laser laparoscopic surgery was introduced in the UK, it was neither clear that training was necessary nor clear who needed it and who should provide it. For some new technologies, the regulatory body, such as the FDA in the USA, will mandate that the manufacturer or retailer provide training as part of the conditions of sale. The extent to which this happens worldwide is hugely variable. Indeed, in developing countries, there is the potential for obsolete developed-world technologies to be introduced with no training offered to local staff.
Clinical engineering as part of health technology management is the primary resource in evaluating problems that have occurred at the device–user interface, the source of most equipment problems. Medical devices should be designed taking into consideration ergonomics and considering possible misuse. Better prepurchase equipment research and evaluation, better techniques to investigate problems that have occurred and improved methods to evaluate systems to prevent future failure have enabled clinical engineers to reduce the risk of medical device failures within healthcare.
Surveillance
Active and passive systems to detect problems with new and existing technologies are paramount to maintaining patient safety. Many device failures will be too infrequent for detection in clinical trials or might only happen in real-world settings sometimes only after several years' use (case study 1). Moreover, not all devices undergo clinical trials prior to introduction. Ideally, there should be a series of surveillance methods to detect problems, as shown in figure 3.
Case study 1 Implants
Some of the most challenging new technologies are implants and materials that are placed in the body permanently. In such technologies, the long-term effects in terms of biocompatibility, durability and non-toxicity of the materials need careful consideration when considering safety. Problems and failures might become evident only after several years, but safety data prior to introduction tend only to be gathered for a short period of time. For example, the long-term problems with hip-replacement prostheses, where the endurance of the fixation materials failed, only occurred after several years of use. Similarly, drug-eluting stents for coronary arteries are only now, after several years of widespread use, known to be associated with increased risk of late thrombotic complications. This emphasises the need for technologies and implants that deal with the vital functions (eg, cardiac pacemakers or vascular stents/prostheses) to be evaluated by a thorough safety appraisal prior to widespread use, as well as ensuring long-term surveillance.
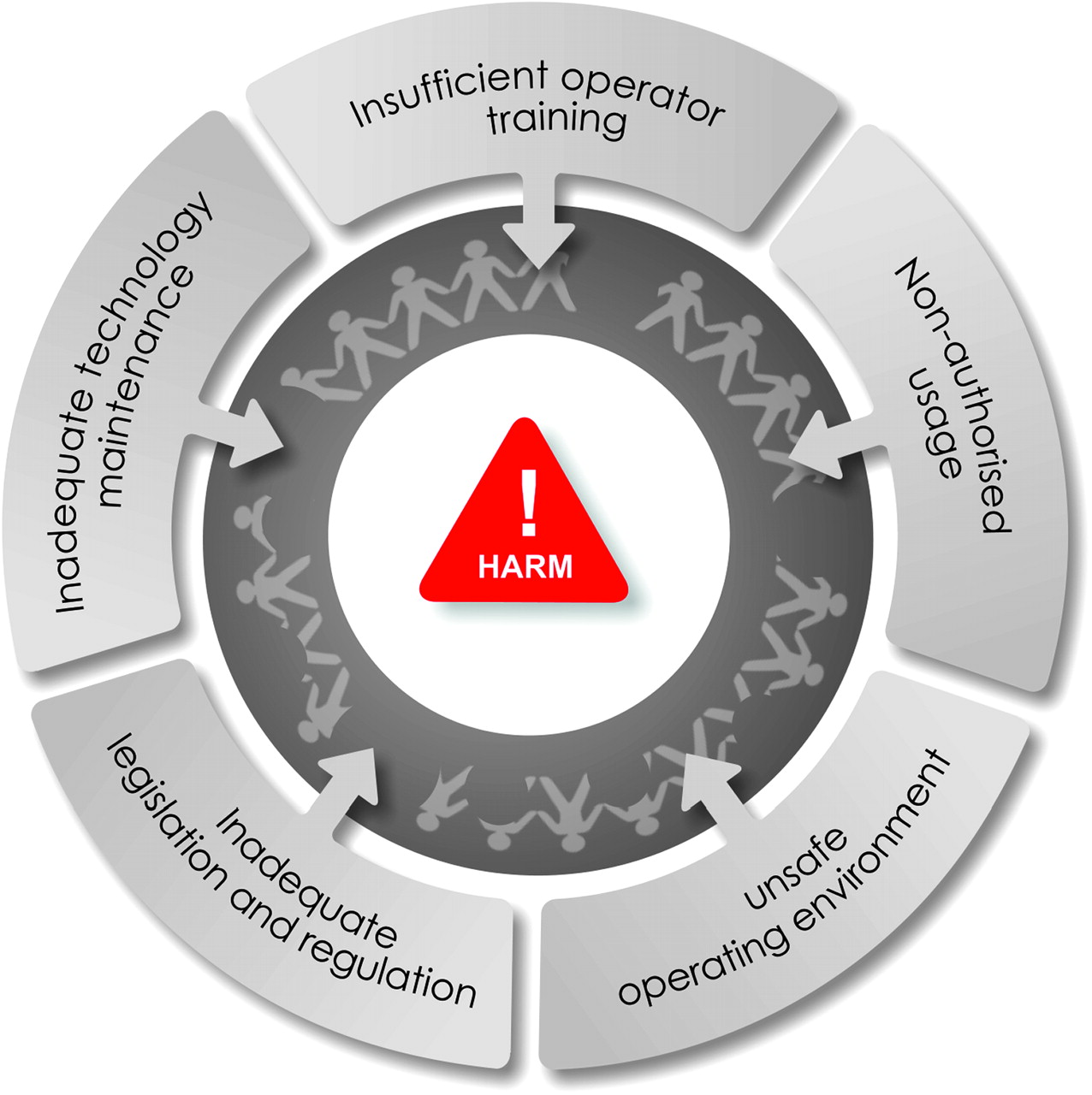
{kind=link}
{kind=link}
{kind=link}
Factors contributing to ‘unsafe’ technology in developing countries. A variety of factors can contribute in isolation or combination to make ‘safe’ developed world technology unsafe in the developing world.
The UK has several such overlapping systems. The Medicines and Healthcare Products Regulatory Agency (MHRA) collects reports on problems with medical devices from patients, healthcare practitioners and manufacturers. Around 8000 reports each year are collected; in 2007, this led to 79 alerts being issued, some of which were serious enough to warrant immediate action.13 Also, in the UK, 23 700 reports concerning the safety of medical devices were collected by the National Patient Safety Agency in the same year.14 A further source of information is the General Practice Research Database, a database containing the anonymous records on patients registered at over 480 general practices in the UK, and also managed by the MHRA.5 This database can be used to monitor the safety and risk–benefit ratio of new medicines and devices, although in practice it is mostly used for monitoring new medicines. The MHRA also proactively tests medical devices.
Many countries, particularly developing countries, have no organised system for reporting, detection, inspection or regulation of issues connected with performance of medical devices and equipment, let alone for tracking safety issues. Encouragingly, more countries are beginning to develop systems. In India, for example, a proposal for an Indian Medical Devices Regulatory Authority (MDRA) has recently been accepted and will be shortly enacted in legislation. The Indian MDRA also intends that devices and equipment conform to applicable global medical device standards in terms of safety and performance, in keeping with the principle of harmonisation. The regulation might provide for mandatory reporting by the manufacturers and voluntary reporting by users or other interested persons or organisations of such events.
Device surveillance or ‘reporting and learning systems’ might be good at identifying potential risk areas or ‘risk signals,’ but the lack of a clear denominator or comparator group makes judgement about ‘increased’ risk due to the device as opposed to the illness itself hard. The UK National Patient Safety Agency issued an alert based on a high rate of sudden death associated with cement use in hemiarthroplasty and arthroplasty,15 but the ability to accurately estimate the increased risk was limited by the lack of a denominator. Once an increased risk has been proven, a further judgement about acceptable risks associated with treatment might also need to be made. Some forms of harm, such as psychological harm, can be hard to assess, whereas others like radiation might not become apparent until years after exposure. Observational studies might be a preferred method to assess harm but are often criticised for potential bias.
Care and thought needs to be given not only to establishing surveillance systems but to what data are collected, how they are analysed and how this information is fed back to manufacturers and users. Device-learning systems not only highlight which devices are failing and how, but also show a significant proportion of hazardous events related to inappropriate use of the technology. Electronic records offer tremendous potential for surveillance in the future. Pharmacovigilance systems are already moving in this direction. Linking unique implant numbers, or device numbers, to patients has tremendous potential for ongoing surveillance in the future.
Situation in the developing world
A variety of factors affect the introduction of new technology in the developing world (see box 2). Many documents have been developed to guide governments, non-governmental agencies, facilities and individuals in managing new technologies safely and correctly in the developing world. However, there is little evidence of capacity development, and compliance with advice remains a problem. Improvement will require significant resources, political will and expertise.
Box 2 Some special considerations in the developing world
Environmental considerations tend to limit the type and range of technologies that can be introduced and the lifespan of a technology. In some instances, premature device failure (eg, an oxygen saturation probe giving a falsely high reading as it degrades) might prevent or delay an appropriate response to prevent patient harm. More specifically, the hospital environment—overheating, poor maintenance of electrical circuits and the presence of vermin—might threaten patient safety. Similarly, the infrastructure, such as poor transport networks, can hinder attempts to maintain and repair new technology in a timely and costly manner.
Inadequate legislation and inconsistent levels of national standards, health technology policy and health professional governance means that there are weak mechanisms to guide and enforce the introduction of new technologies. A vendor can sell an inappropriate or unsafe device, and health professionals (or non-health professionals) can use it without any recourse, oversight or correction in many countries.
Lack of finance or financial governance means there are limited funds to procure devices, maintain devices (including restocking of reagents and disposable parts) and train staff adequately. Many new technologies depend on disposables and reagents. The costs and logistics of providing these means that technologies are often used outside their design specifications, which can lead to device failure and patient harm.
Different skills and knowledge among healthcare professionals, as compared with developed countries where the technology was established, might hinder the safe introduction of new technologies. Technologies are acquired or thrust upon developing countries without an understanding of the need or capacity of the areas to absorb them. The technologies can cause harm if used incorrectly (or used by individuals who are not trained), installed incorrectly or serviced incorrectly. Capacity development and education are key to ensuring safe technologies.
In the past, new technology in the developing world often meant old, obsolete or non-efficacious technologies from the developed world being donated. These technologies were generally introduced without any evaluation process, although their impact on patient safety might be less because of limited use. Most of these donated or sold technologies were non-functional at introduction or inappropriate for use and were often discarded or stored at the back of hospitals. In Colombia, it was estimated that up to 96% of foreign-donated technologies stopped functioning within 5 years of being donated, and that 39% never worked at all.16 Even when new modern technologies are introduced (see case study 2), careful thought should be given to exactly how they are introduced and the extent to which they are appropriate for the setting; otherwise they could introduce new harms.
Case study 2 Laboratory technology in the developing world
Modern haematology analysers and CD4+ T cell counters are being introduced to provide antiretroviral drug treatment for HIV patients. These platforms require clean areas, high skills, in date reagents, in date controls and cold chain maintenance, among other factors. In developing-world settings, the safety risk of assuming that the haematology analyser is right or the CD4+ T cell counter unit is correct is significant. There are more basic and nearly as efficacious methods of providing the information to guide drug dosing—for example, the use of dyna-bead or dipstick-style low-tech direct measures of CD4+ T cell counts. The importance of appropriate technology, tailored to local needs (environmental, ethical, cultural, social and economic), should be considered in these settings.
A variety of factors can work together or alone to make ‘safe’ technology in the developed world become unsafe. There is a clear need for appropriate technologies to be developed for and introduced to such settings. It is important not to become so focused on basic technologies as ‘appropriate technologies’ that opportunities are missed for acceptable technology jumps, so-called leapfrog technologies.17 Cellular phones provide an example where a technology jump in the developing world has improved the communication capacity of many of the world's poorest citizens. Moreover, with increasing high-level funding of health programmes, there could be a new trend for introducing new and often cutting-edge technologies to the developing world. As such ‘appropriate technologies’ tend to be efficient, reliable and easier to use, they might actually find a market in the developed world, particularly where they can under cut more expensive technologies. Although some people suggest ‘appropriate technology’ is inferior, there is a strong case for using ‘appropriate technologies’ in preference to cutting-edge technologies in many developing countries (case study 2), at least until healthcare and administrative systems are sufficiently developed to handle all aspects associated with new technologies.
Policy decisions
The more scarce the resources, the more rational should be the decisions made about the acquisition and utilisation of any technology. This is now true not only for developing countries, as it used to be the priority in the past, but also for any country in the world. Cost is an issue today more than ever. This is one of the main reasons why health technology assessment is absolutely necessary. In addition, in recent years it has become clear that not all technologies are safe and can cause harm to patients if they are not introduced correctly within healthcare systems or if they are utilised inappropriately.
There are pressures to introduce new technologies without adequate assessment. One of the pressures is, of course, the pace of technology development itself. The faster the development of new technology, the harder it is to maintain an appropriate speed for making timely assessments before their utilisation. The risk for the patients is then high. Pressure is also posed by the manufacturers who have a strong incentive to promote and sell their products as soon as possible. Another pressure is created by healthcare professionals, particularly by doctors who are permanently inclined to have state-of-the-art technologies, even though they might not be as cost-effective as the technology they already utilise. Therefore, between the vested interests of manufacturers and the increasing interest of doctors, there is frequently a vacuum of information on cost-effectiveness and patient safety for the most advanced technology. In these situations, introducing technology safely is an imperative to rational decision-making not only to save money but also to ensure value for money, mainly in terms of patient safety.
The factors that create pressures and their consequences are equally applicable, whether technology is bought by private or public providers. Therefore, there is a need for an independent agency to produce information for better decision-making, either for private consumers or for governments when the latter finance the acquisition of technology. Governments can then act not only as finance agencies, but also as regulators and evaluators of technology to introduce it safely into healthcare systems in order to protect patients from potential harm. To achieve this aim, reliable and appropriate data are required. Moreover, the professionals must be able to interpret the data to produce useful and timely information for decision-making.
Conclusions
New technology, both conceptually and contextually new, introduces risks that are often unforeseen. Regulation, health technology assessment and clinical engineering, together with staff training, have a vital role in ensuring the safety of new technologies. Information will often be imperfect, emphasising the need for ongoing systems of surveillance, which should be multifaceted. Knowledge about safety will often be much better for old technologies than for conceptually new ones. The challenge is often introducing such systems efficiently to promote safety, while minimising bureaucracy. Some gains might be made by harmonisation of decision-making processes, but inevitably some decisions will need to be tailored to national or local needs. The developing world faces particular challenges, lacking both a regulatory structure and sufficient resources. The use of redundant or inappropriate technologies established in the developing world rather than ‘appropriate technologies’ further exacerbates these problems. Safety dimensions, including safety under real-world conditions, must be part of the decision-making process on the introduction of new technologies into health systems by governments, insurers, donor agencies, financing agencies and UN agencies. Patients, healthcare professionals and decision-makers should be aware that new technologies are not always better than existing ones.
Footnotes
Linked articles 040428
Funding The project was funded by WHO Patient Safety.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Original research